

Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome
- PMID: 24352790
- PMCID: PMC3976336
- DOI: 10.1093/hmg/ddt640
Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome
Erratum in
- Hum Mol Genet. 2014 Dec 15;23(24):6695
Abstract
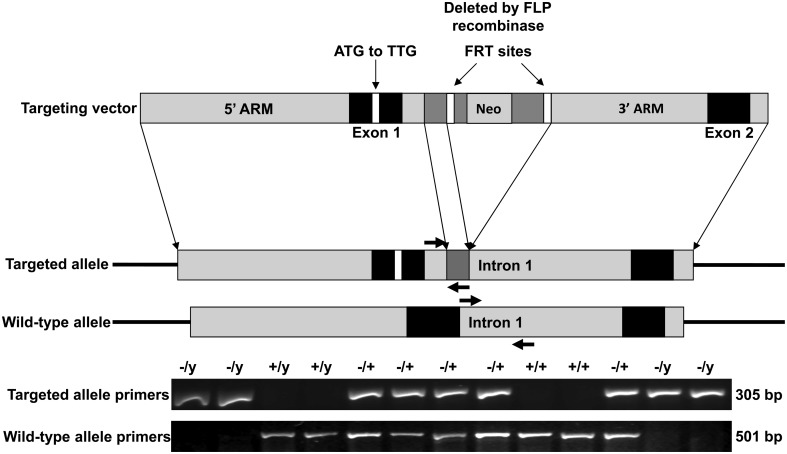
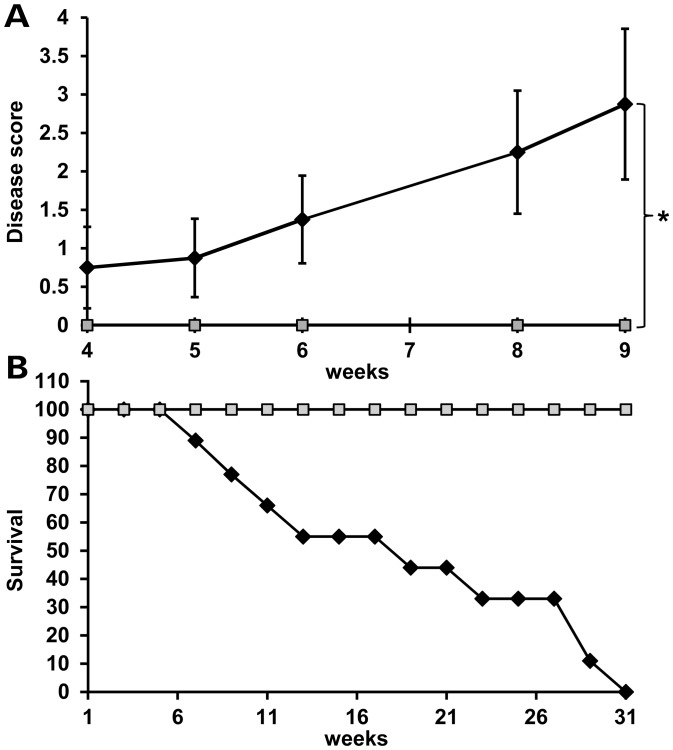
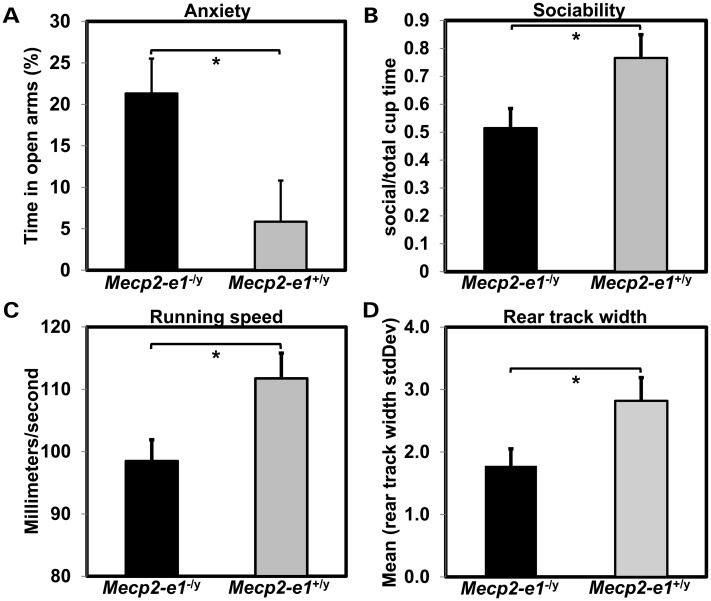
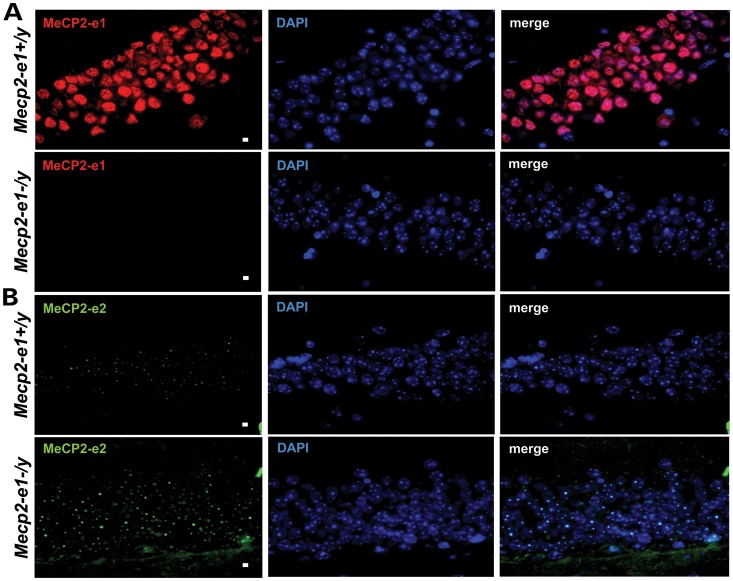
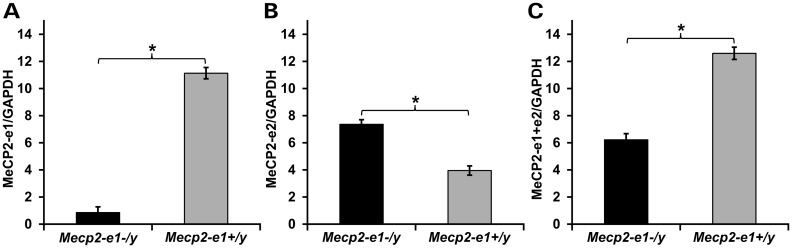
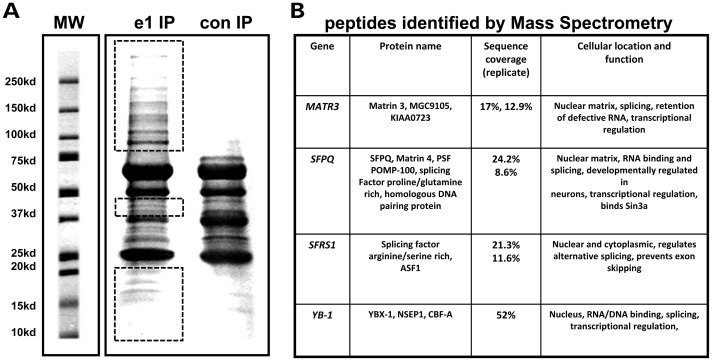
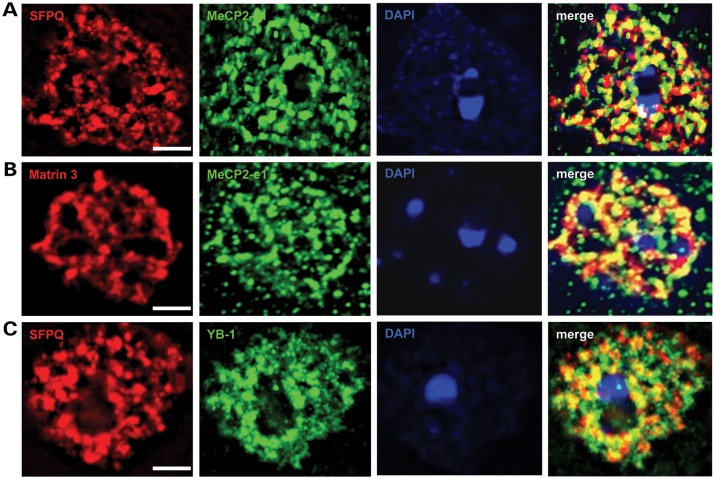
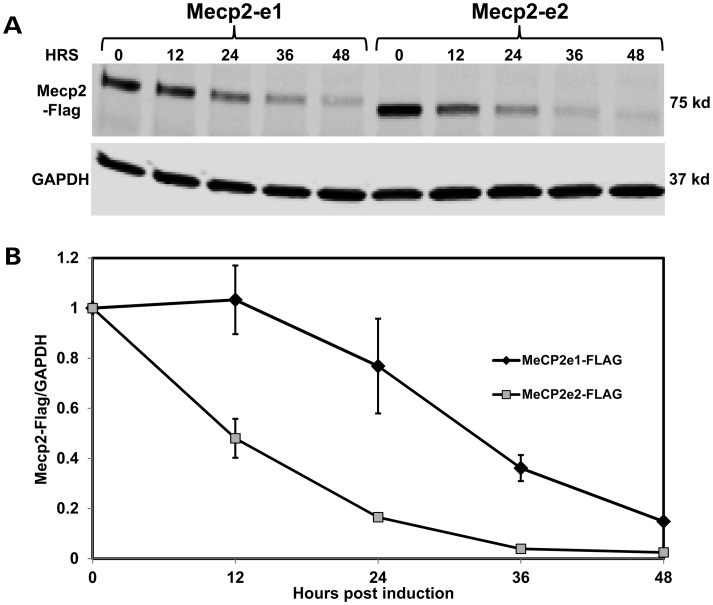
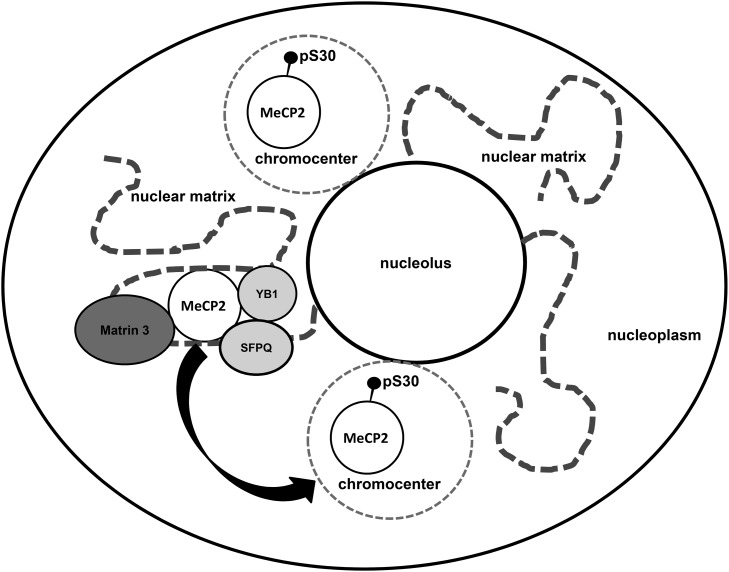
Mutations in MECP2 cause the neurodevelopmental disorder Rett syndrome (RTT OMIM 312750). Alternative inclusion of MECP2/Mecp2 exon 1 with exons 3 and 4 encodes MeCP2-e1 or MeCP2-e2 protein isoforms with unique amino termini. While most MECP2 mutations are located in exons 3 and 4 thus affecting both isoforms, MECP2 exon 1 mutations but not exon 2 mutations have been identified in RTT patients, suggesting that MeCP2-e1 deficiency is sufficient to cause RTT. As expected, genetic deletion of Mecp2 exons 3 and/or 4 recapitulates RTT-like neurologic defects in mice. However, Mecp2 exon 2 knockout mice have normal neurologic function. Here, a naturally occurring MECP2 exon 1 mutation is recapitulated in a mouse model by genetic engineering. A point mutation in the translational start codon of Mecp2 exon 1, transmitted through the germline, ablates MeCP2-e1 translation while preserving MeCP2-e2 production in mouse brain. The resulting MeCP2-e1 deficient mice developed forelimb stereotypy, hindlimb clasping, excessive grooming and hypo-activity prior to death between 7 and 31 weeks. MeCP2-e1 deficient mice also exhibited abnormal anxiety, sociability and ambulation. Despite MeCP2-e1 and MeCP2-e2 sharing, 96% amino acid identity, differences were identified. A fraction of phosphorylated MeCP2-e1 differed from the bulk of MeCP2 in subnuclear localization and co-factor interaction. Furthermore, MeCP2-e1 exhibited enhanced stability compared with MeCP2-e2 in neurons. Therefore, MeCP2-e1 deficient mice implicate MeCP2-e1 as the sole contributor to RTT with non-redundant functions.
Figures
References
-
- Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. - PubMed
-
- Coy J.F., Sedlacek Z., Bachner D., Delius H., Poustka A. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3″-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum. Mol. Genet. 1999;8:1253–1262. - PubMed
-
- Mnatzakanian G.N., Lohi H., Munteanu I., Alfred S.E., Yamada T., MacLeod P.J., Jones J.R., Scherer S.W., Schanen N.C., Friez M.J., et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004;36:339–341. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
