

Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease
- PMID: 24354792
- PMCID: PMC3952797
- DOI: 10.1111/bph.12544
Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease
Abstract
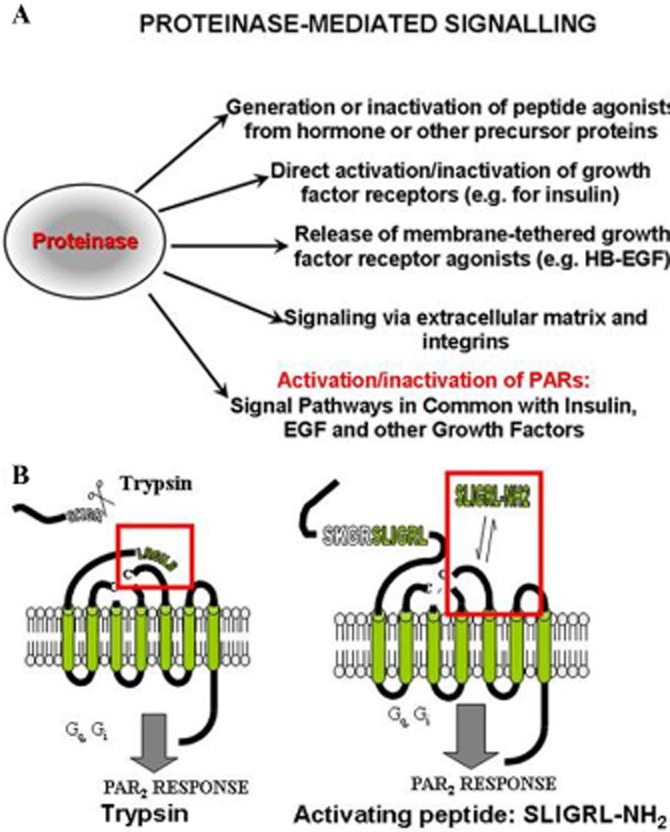
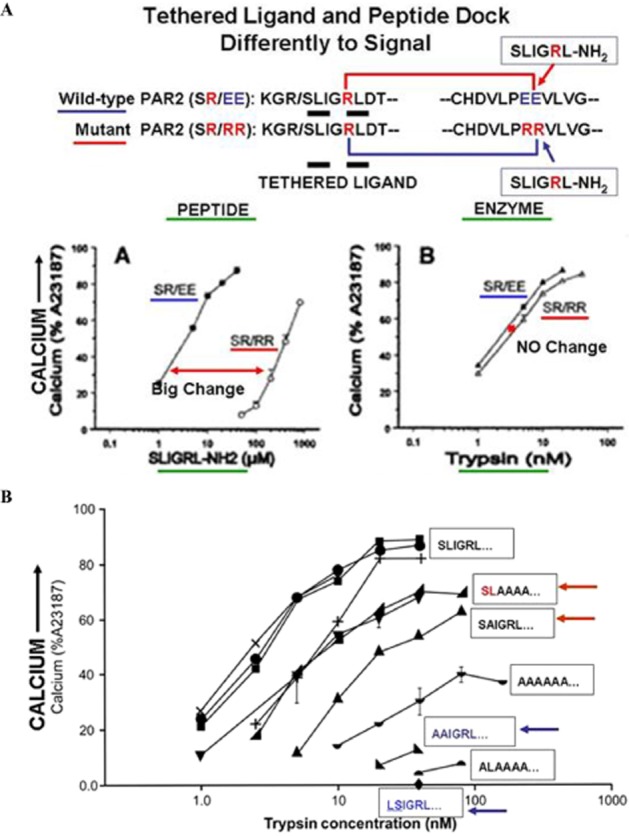
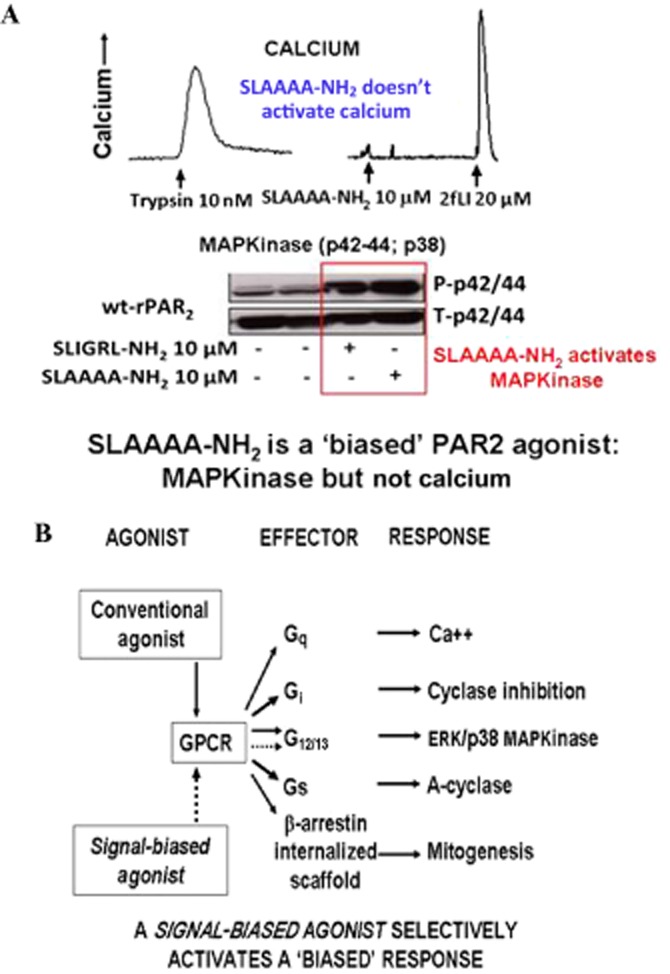
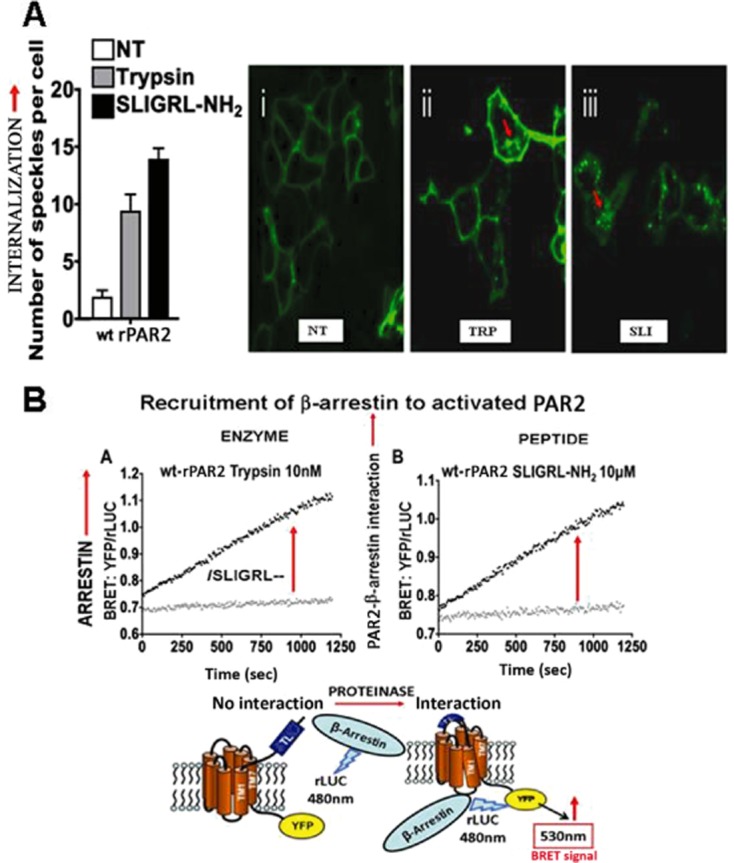
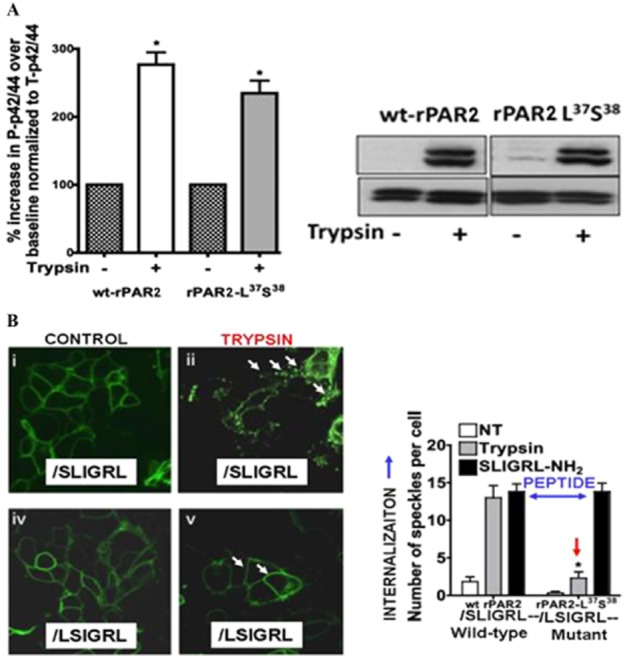
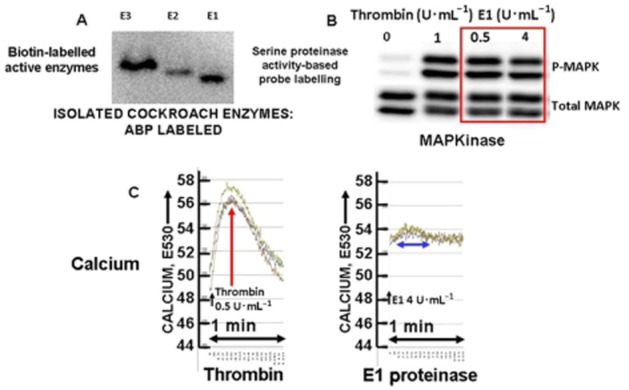
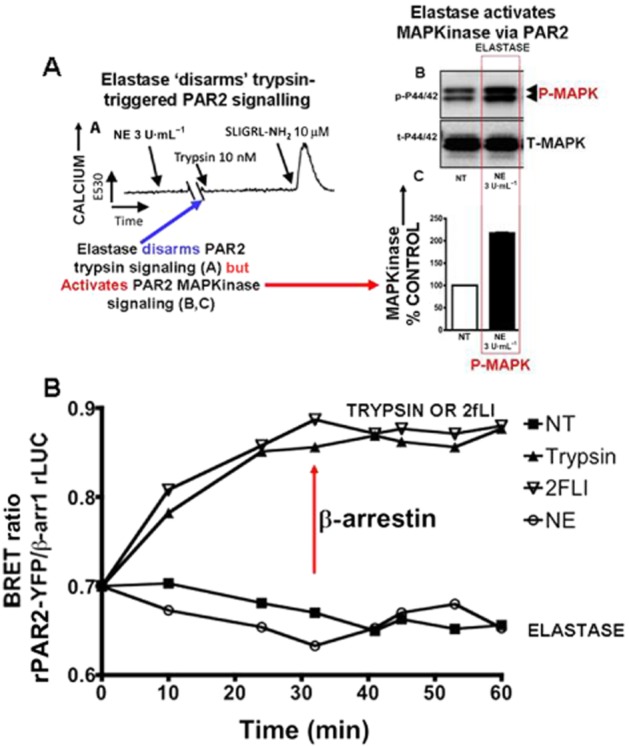
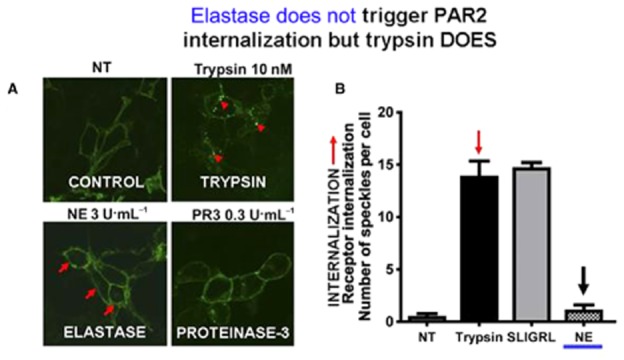
Although it has been known since the 1960s that trypsin and chymotrypsin can mimic hormone action in tissues, it took until the 1990s to discover that serine proteinases can regulate cells by cleaving and activating a unique four-member family of GPCRs known as proteinase-activated receptors (PARs). PAR activation involves the proteolytic exposure of its N-terminal receptor sequence that folds back to function as a 'tethered' receptor-activating ligand (TL). A key N-terminal arginine in each of PARs 1 to 4 has been singled out as a target for cleavage by thrombin (PARs 1, 3 and 4), trypsin (PARs 2 and 4) or other proteases to unmask the TL that activates signalling via Gq , Gi or G12 /13 . Similarly, synthetic receptor-activating peptides, corresponding to the exposed 'TL sequences' (e.g. SFLLRN-, for PAR1 or SLIGRL- for PAR2) can, like proteinase activation, also drive signalling via Gq , Gi and G12 /13 , without requiring receptor cleavage. Recent data show, however, that distinct proteinase-revealed 'non-canonical' PAR tethered-ligand sequences and PAR-activating agonist and antagonist peptide analogues can induce 'biased' PAR signalling, for example, via G12 /13 -MAPKinase instead of Gq -calcium. This overview summarizes implications of this 'biased' signalling by PAR agonists and antagonists for the recognized roles the PARs play in inflammatory settings.
Keywords: GPCR; activated protein-C; biased signalling; protease; proteinase-activated receptor; thrombin.
© 2013 The British Pharmacological Society.
Figures
References
-
- Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130:248–282. - PubMed
-
- Al-Ani B, Saifeddine M, Hollenberg MD. Detection of functional receptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can J Physiol Pharmacol. 1995;73:1203–1207. - PubMed
-
- Al-Ani B, Wijesuriya SJ, Hollenberg MD. Proteinase-activated receptor 2: differential activation of the receptor by tethered ligand and soluble peptide analogs. J Pharmacol Exp Ther. 2002;302:1046–1054. - PubMed
-
- Al-Ani B, Hansen KK, Hollenberg MD. Proteinase-activated receptor-2: key role of amino-terminal dipeptide residues of the tethered ligand for receptor activation. Mol Pharmacol. 2004;65:149–156. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
