

(R,R')-4'-methoxy-1-naphthylfenoterol targets GPR55-mediated ligand internalization and impairs cancer cell motility
- PMID: 24355564
- PMCID: PMC3935314
- DOI: 10.1016/j.bcp.2013.11.020
(R,R')-4'-methoxy-1-naphthylfenoterol targets GPR55-mediated ligand internalization and impairs cancer cell motility
Abstract
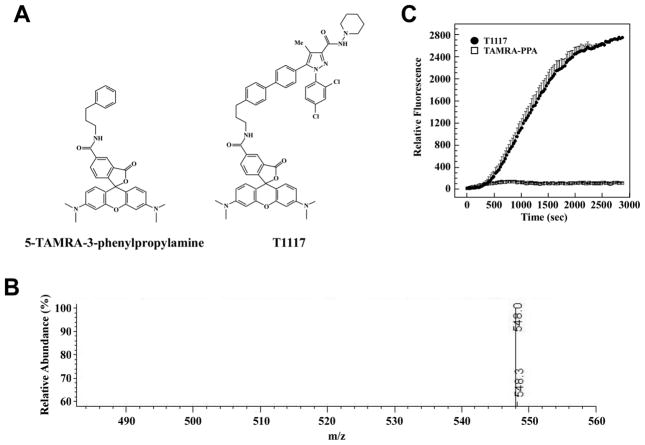
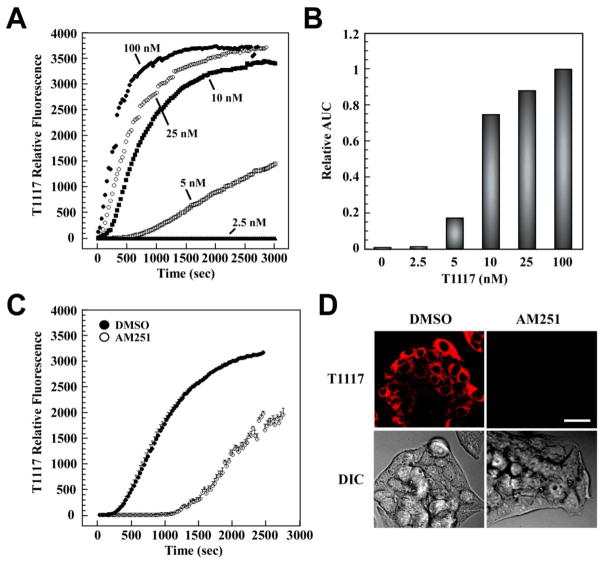
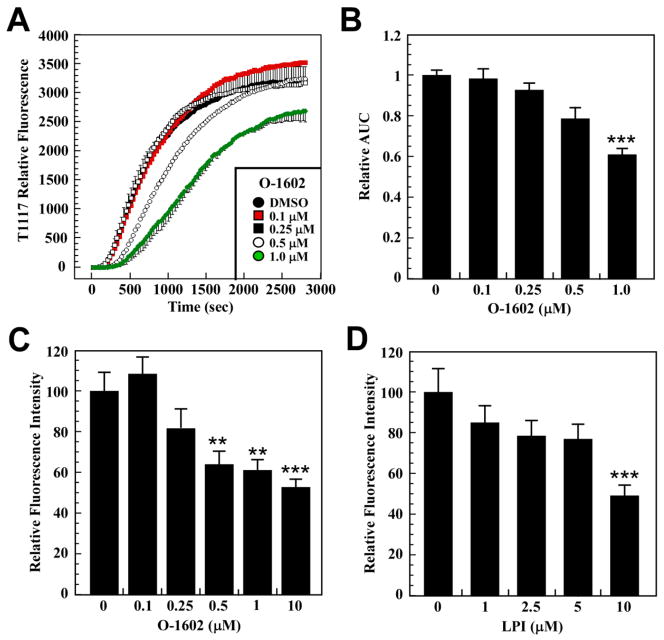
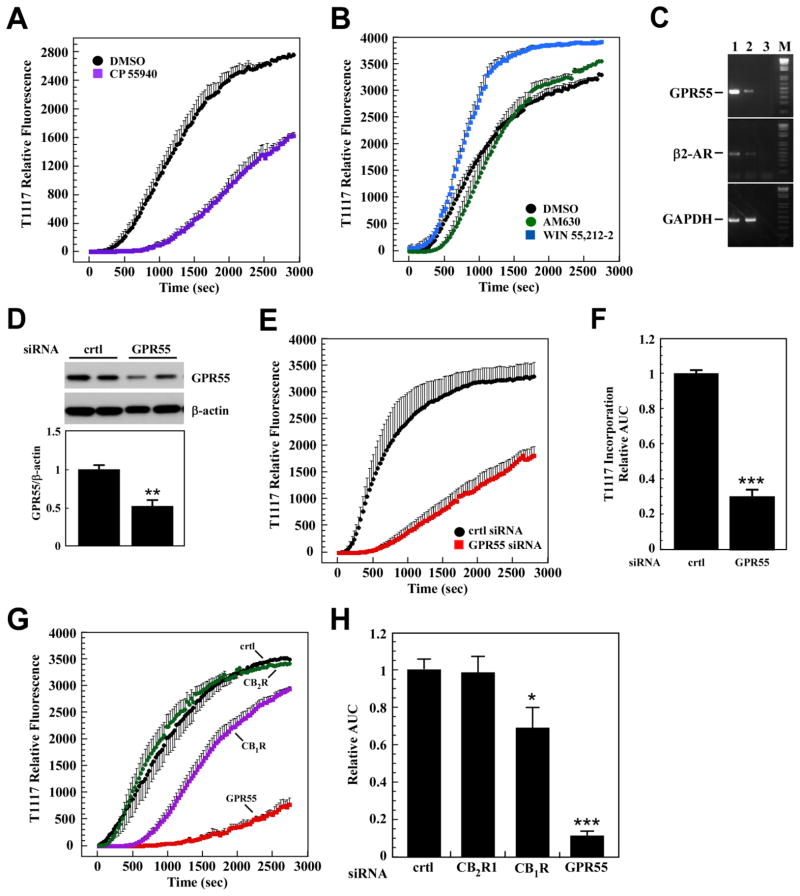
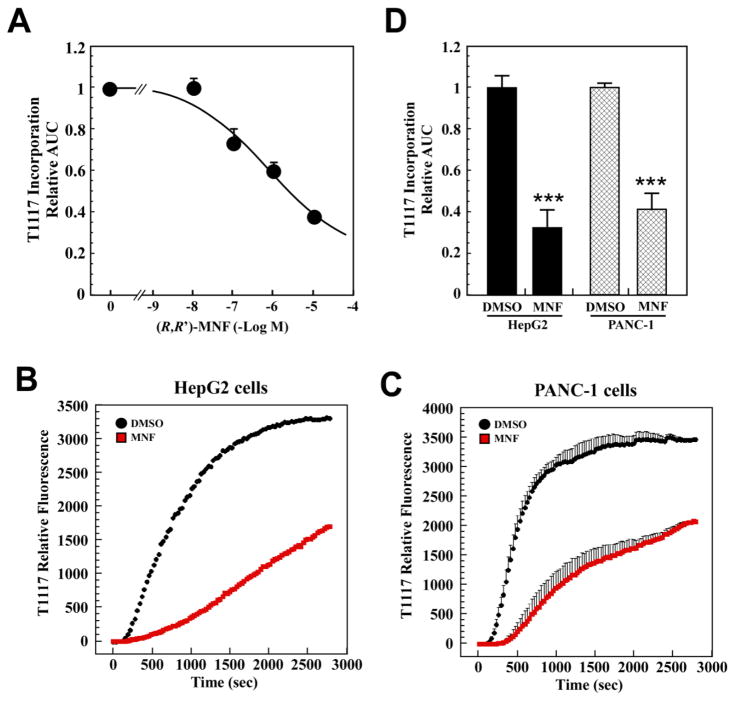
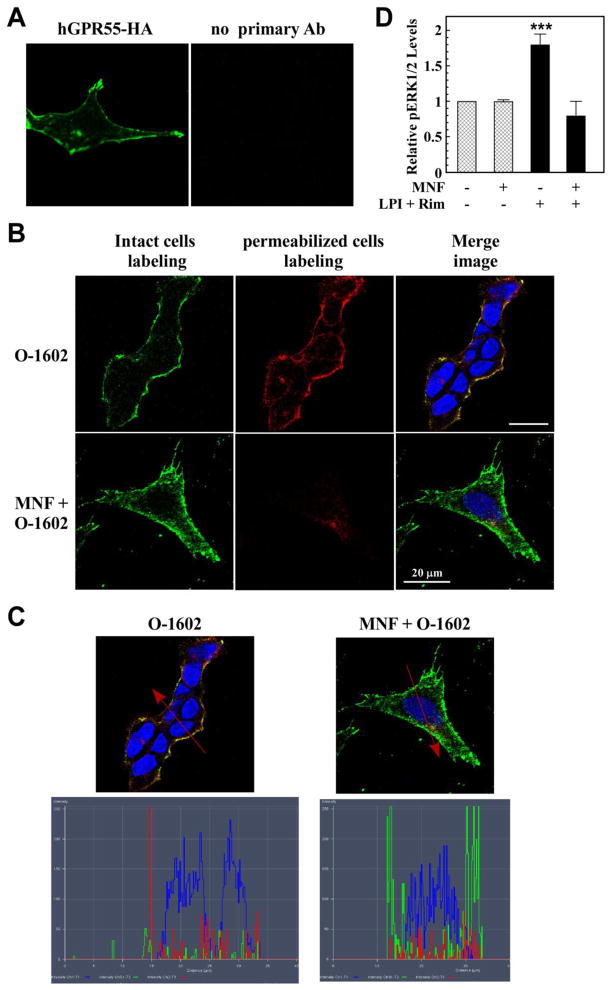
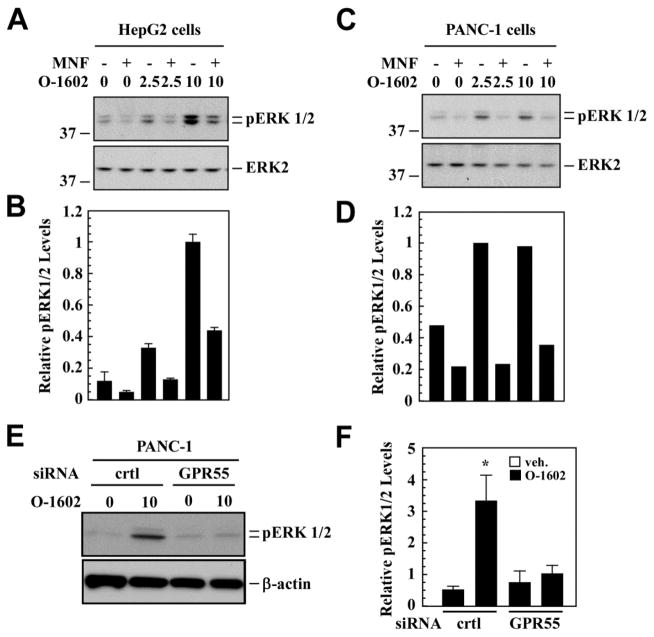
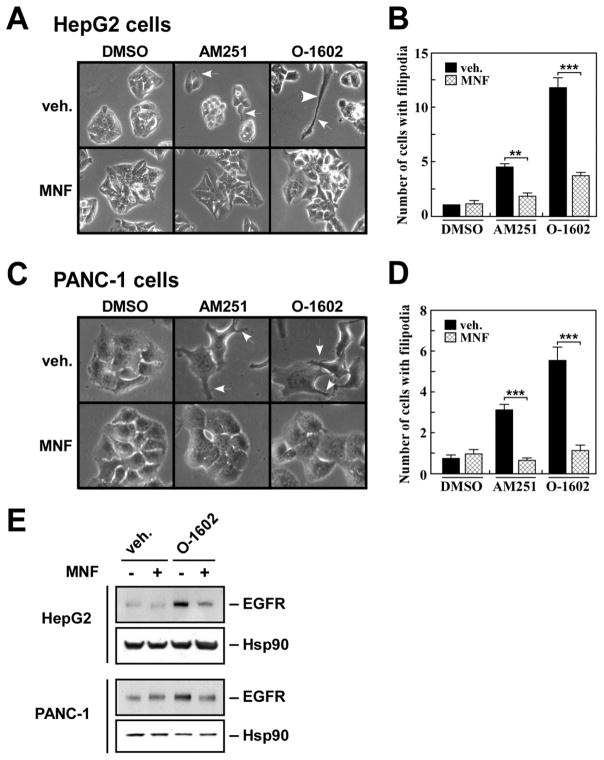
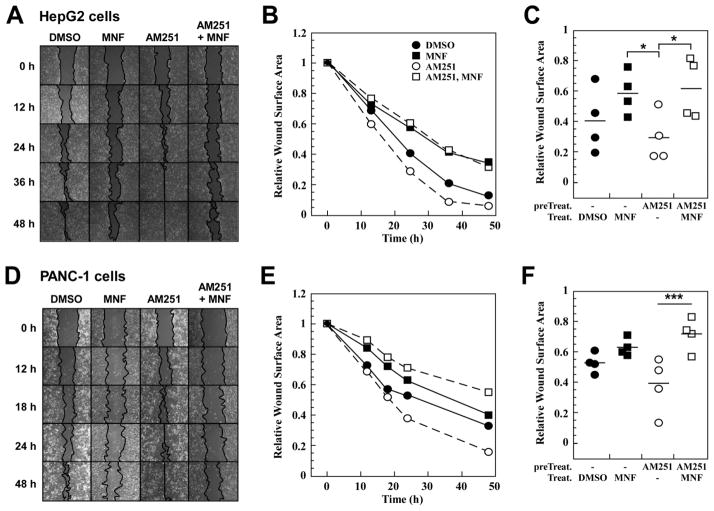
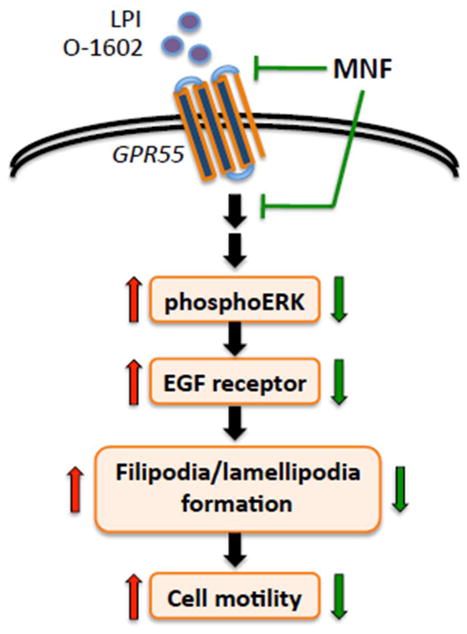
(R,R')-4'-Methoxy-1-naphthylfenoterol (MNF) promotes growth inhibition and apoptosis of human HepG2 hepatocarcinoma cells via cannabinoid receptor (CBR) activation. The synthetic CB1R inverse agonist, AM251, has been shown to block the anti-mitogenic effect of MNF in these cells; however, AM251 is also an agonist of the recently deorphanized, lipid-sensing receptor, GPR55, whose upregulation contributes to carcinogenesis. Here, we investigated the role of MNF in GPR55 signaling in human HepG2 and PANC-1 cancer cell lines in culture by focusing first on internalization of the fluorescent ligand Tocrifluor 1117 (T1117). Initial results indicated that cell pretreatment with GPR55 agonists, including the atypical cannabinoid O-1602 and l-α-lysophosphatidylinositol, dose-dependently reduced the rate of cellular T1117 uptake, a process that was sensitive to MNF inhibition. GPR55 internalization and signaling mediated by O-1602 was blocked by MNF in GPR55-expressing HEK293 cells. Pretreatment of HepG2 and PANC-1 cells with MNF significantly abrogated the induction of ERK1/2 phosphorylation in response to AM251 and O-1602. Moreover, MNF exerted a coordinated negative regulation of AM251 and O-1602 inducible processes, including changes in cellular morphology and cell migration using scratch wound healing assay. This study shows for the first time that MNF impairs GPR55-mediated signaling and, therefore, may have therapeutic potential in the management of cancer.
Keywords: Cell motility; Cellular morphology; G-protein coupled receptor; GPR55; Ligand internalization.
Published by Elsevier Inc.
Figures
Similar articles
-
Selective GPR55 antagonism reduces chemoresistance in cancer cells.Pharmacol Res. 2016 Sep;111:757-766. doi: 10.1016/j.phrs.2016.07.013. Epub 2016 Jul 14. Pharmacol Res. 2016. PMID: 27423937 Free PMC article.
-
Concurrent activation of β2-adrenergic receptor and blockage of GPR55 disrupts pro-oncogenic signaling in glioma cells.Cell Signal. 2017 Aug;36:176-188. doi: 10.1016/j.cellsig.2017.05.006. Epub 2017 May 8. Cell Signal. 2017. PMID: 28495590 Free PMC article.
-
Cannabinoid receptor activation correlates with the proapoptotic action of the β2-adrenergic agonist (R,R')-4-methoxy-1-naphthylfenoterol in HepG2 hepatocarcinoma cells.J Pharmacol Exp Ther. 2012 Oct;343(1):157-66. doi: 10.1124/jpet.112.195206. Epub 2012 Jul 9. J Pharmacol Exp Ther. 2012. PMID: 22776956 Free PMC article.
-
Pharmacological characterization of GPR55, a putative cannabinoid receptor.Pharmacol Ther. 2010 Jun;126(3):301-13. doi: 10.1016/j.pharmthera.2010.02.004. Epub 2010 Mar 16. Pharmacol Ther. 2010. PMID: 20298715 Free PMC article. Review.
-
GPR55, a lysophosphatidylinositol receptor with cannabinoid sensitivity?Curr Top Med Chem. 2010;10(8):799-813. doi: 10.2174/156802610791164229. Curr Top Med Chem. 2010. PMID: 20370712 Review.
Cited by
-
Antitumor Cannabinoid Chemotypes: Structural Insights.Front Pharmacol. 2019 May 31;10:621. doi: 10.3389/fphar.2019.00621. eCollection 2019. Front Pharmacol. 2019. PMID: 31214034 Free PMC article.
-
GPR55 is expressed in glutamate neurons and functionally modulates drug taking and seeking in rats and mice.Transl Psychiatry. 2024 Feb 19;14(1):101. doi: 10.1038/s41398-024-02820-3. Transl Psychiatry. 2024. PMID: 38374108 Free PMC article.
-
Deprogramming metabolism in pancreatic cancer with a bi-functional GPR55 inhibitor and biased β2 adrenergic agonist.Sci Rep. 2022 Mar 7;12(1):3618. doi: 10.1038/s41598-022-07600-x. Sci Rep. 2022. PMID: 35256673 Free PMC article.
-
Selective GPR55 antagonism reduces chemoresistance in cancer cells.Pharmacol Res. 2016 Sep;111:757-766. doi: 10.1016/j.phrs.2016.07.013. Epub 2016 Jul 14. Pharmacol Res. 2016. PMID: 27423937 Free PMC article.
-
The Mechanisms of GPR55 Receptor Functional Selectivity during Apoptosis and Proliferation Regulation in Cancer Cells.Int J Mol Sci. 2023 Mar 14;24(6):5524. doi: 10.3390/ijms24065524. Int J Mol Sci. 2023. PMID: 36982628 Free PMC article.
References
-
- Jozwiak K, Khalid C, Tanga MJ, Berzetei-Gurske I, Jimenez L, Kozocas JA, et al. Comparative molecular field analysis of the binding of the stereoisomers of fenoterol and fenoterol derivatives to the beta2 adrenergic receptor. J Med Chem. 2007;50:2903–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
