

ERO1α-dependent endoplasmic reticulum-mitochondrial calcium flux contributes to ER stress and mitochondrial permeabilization by procaspase-activating compound-1 (PAC-1)
- PMID: 24357799
- PMCID: PMC3877569
- DOI: 10.1038/cddis.2013.502
ERO1α-dependent endoplasmic reticulum-mitochondrial calcium flux contributes to ER stress and mitochondrial permeabilization by procaspase-activating compound-1 (PAC-1)
Erratum in
-
Correction to: ERO1α-dependent endoplasmic reticulum-mitochondrial calcium flux contributes to ER stress and mitochondrial permeabilization by procaspase-activating compound-1 (PAC-1).Cell Death Dis. 2025 Oct 21;16(1):740. doi: 10.1038/s41419-025-08108-8. Cell Death Dis. 2025. PMID: 41120294 Free PMC article. No abstract available.
Abstract
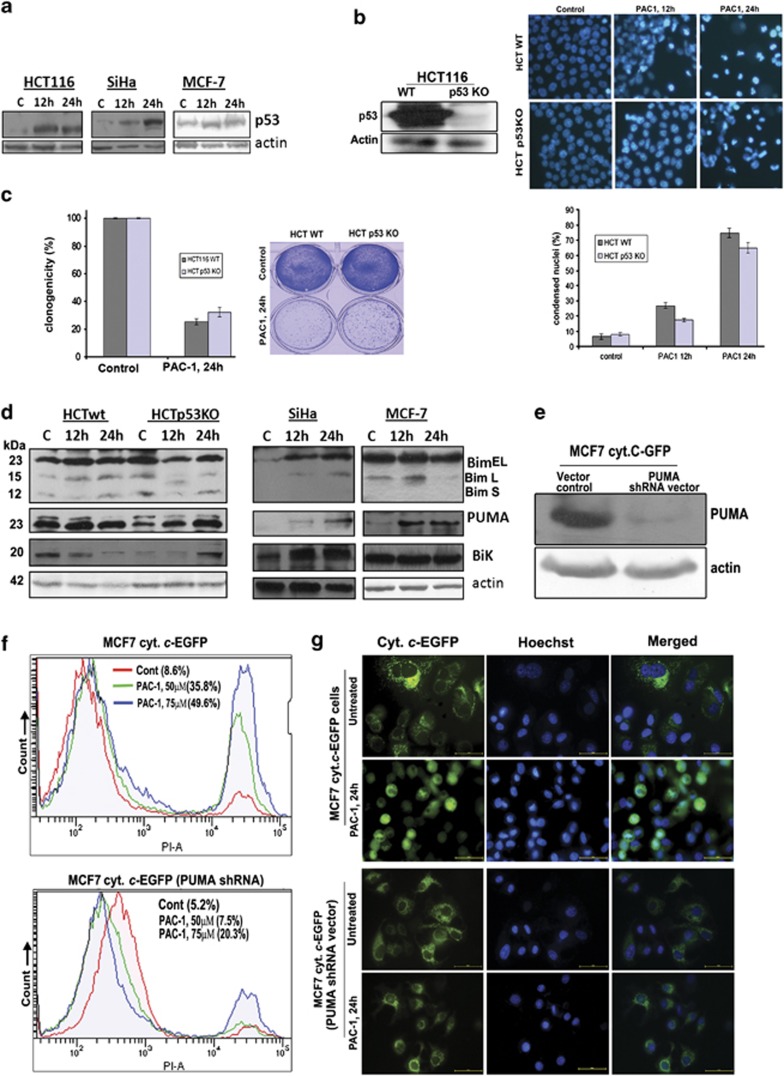
Procaspase-activating compound-1 (PAC-1) is the first direct caspase-activating compound discovered; using an in vitro cell-free system of caspase activation. Subsequently, this compound was shown to induce apoptosis in a variety of cancer cells with promising in vivo antitumor activity in canine lymphoma model. Recently, we have reported its ability to kill drug-resistant, Bcl-2/Bcl-xL overexpressing and Bax/Bak-deficient cells despite the essential requirement of mitochondrial cytochrome c (cyt. c) release for caspase activation, indicating that the key molecular targets of PAC-1 in cancer cells are yet to be identified. Here, we have identified Ero1α-dependent endoplasmic reticulum (ER) calcium leakage to mitochondria through mitochondria-associated ER membranes (MAM) and ER luminal hyper-oxidation as the critical events of PAC-1-mediated cell death. PAC-1 treatment upregulated Ero1α in multiple cell lines, whereas silencing of Ero1α significantly inhibited calcium release from ER and cell death. Loss of ER calcium and hyper-oxidation of ER lumen by Ero1α collectively triggered ER stress. Upregulation of GRP78 and splicing of X-box-binding protein 1 (XBP1) mRNA in multiple cancer cells suggested ER stress as the general event triggered by PAC-1. XBP1 mRNA splicing and GRP78 upregulation confirmed ER stress even in Bax/Bak double knockout and PAC-1-resistant Apaf-1-knockout cells, indicating an induction of ER stress-mediated mitochondrial apoptosis by PAC-1. Furthermore, we identified BH3-only protein p53 upregulated modulator of apoptosis (PUMA) as the key molecular link that orchestrates overwhelmed ER stress to mitochondria-mediated apoptosis, involving mitochondrial reactive oxygen species, in a p53-independent manner. Silencing of PUMA in cancer cells effectively reduced cyt. c release and cell death by PAC-1.
Figures






References
-
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Ann Rev Biochem. 1999;68:383–424. - PubMed
-
- Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. - PubMed
-
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. - PubMed
-
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–699. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
