

Large numbers of individuals are required to classify and define risk for rare variants in known cancer risk genes
- PMID: 24357849
- PMCID: PMC4063879
- DOI: 10.1038/gim.2013.187
Large numbers of individuals are required to classify and define risk for rare variants in known cancer risk genes
Abstract
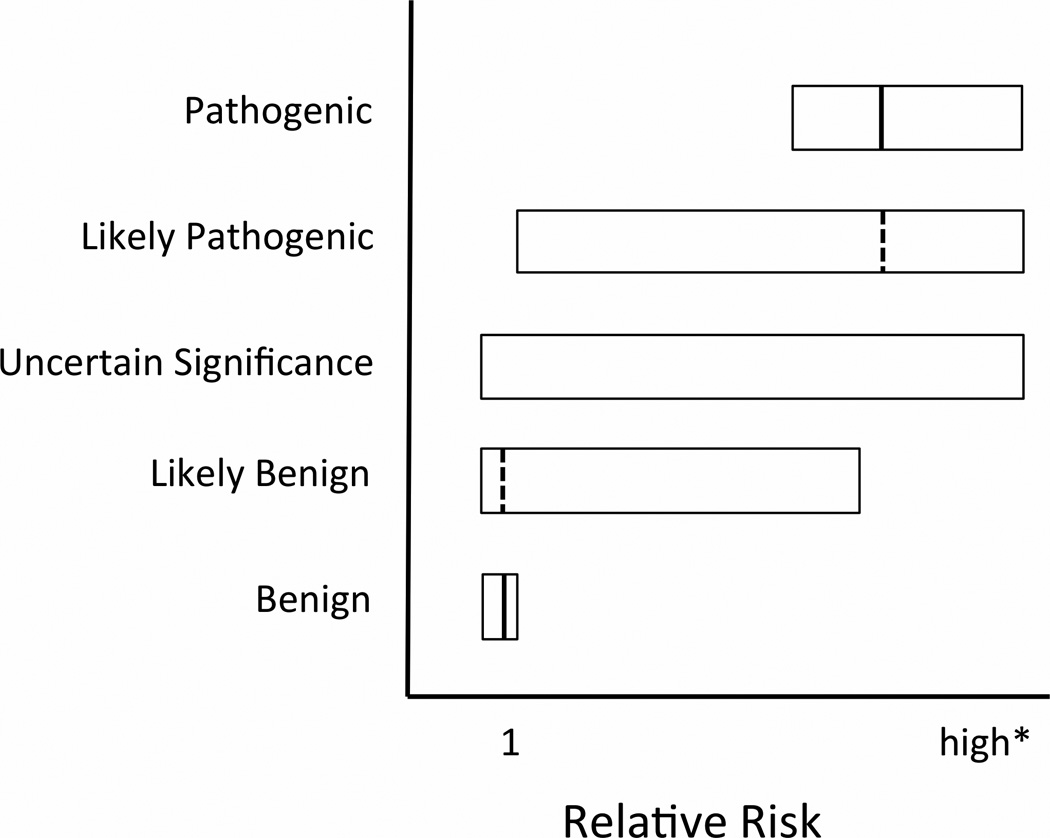
Purpose: Up to half of unique genetic variants in genomic evaluations of familial cancer risk will be rare variants of uncertain significance. Classification of rare variants will be an ongoing issue as genomic testing becomes more common.
Methods: We modified standard power calculations to explore sample sizes necessary to classify and estimate relative disease risk for rare variant frequencies (0.001-0.00001) and varying relative risk (20-1.5), using population-based and family-based designs focusing on breast and colon cancer. We required 80% power and tolerated a 10% false-positive rate because variants tested will be in known genes with high pretest probability.
Results: Using population-based strategies, hundreds to millions of cases are necessary to classify rare cancer variants. Larger samples are necessary for less frequent and less penetrant variants. Family-based strategies are robust to changes in variant frequency and require between 8 and 1,175 individuals, depending on risk.
Conclusion: It is unlikely that most rare missense variants will be classifiable in the near future, and accurate relative risk estimates may never be available for very rare variants. This knowledge may alter strategies for communicating information about variants of uncertain significance to patients.
Figures
References
-
- Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV, Pruss DR, Critchfield GC. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002;20:1480–1490. - PubMed
-
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
