

ERK2-mediated phosphorylation of transcriptional coactivator binding protein PIMT/NCoA6IP at Ser298 augments hepatic gluconeogenesis
- PMID: 24358311
- PMCID: PMC3866170
- DOI: 10.1371/journal.pone.0083787
ERK2-mediated phosphorylation of transcriptional coactivator binding protein PIMT/NCoA6IP at Ser298 augments hepatic gluconeogenesis
Abstract
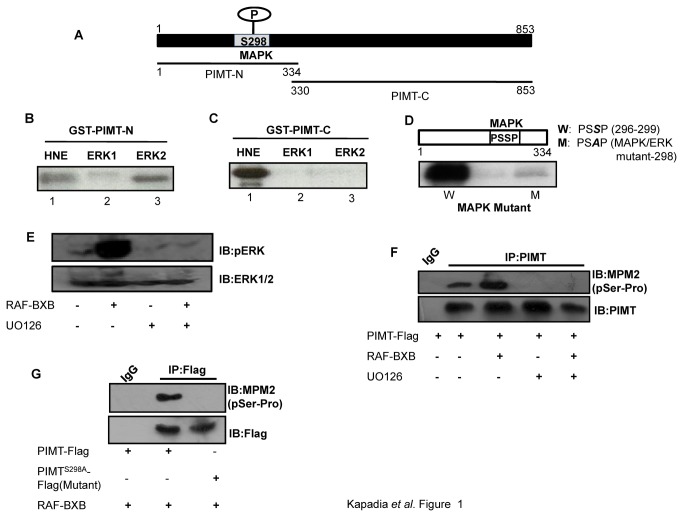
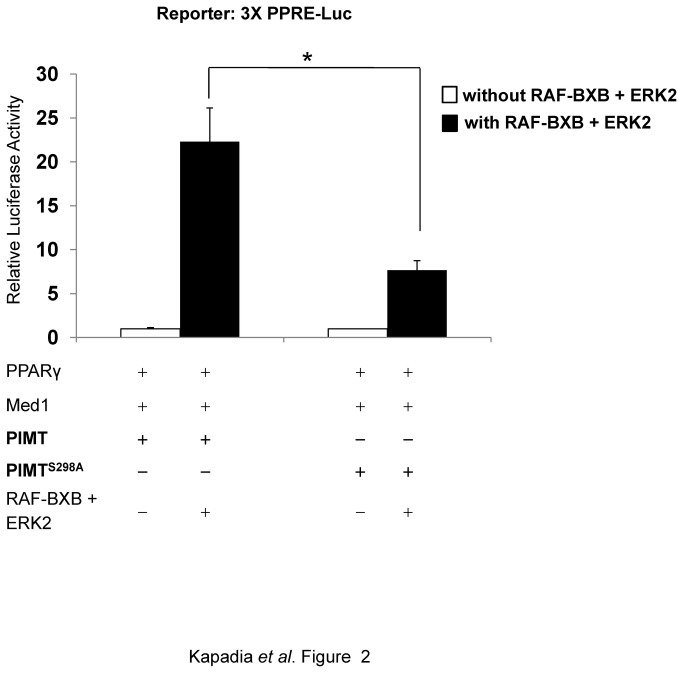
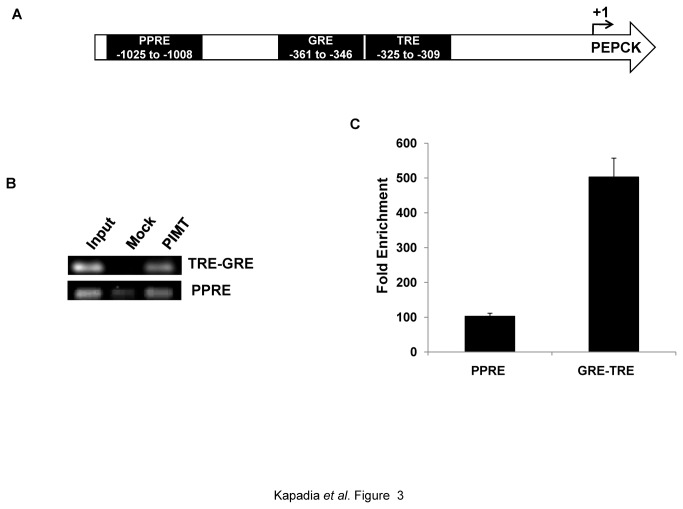
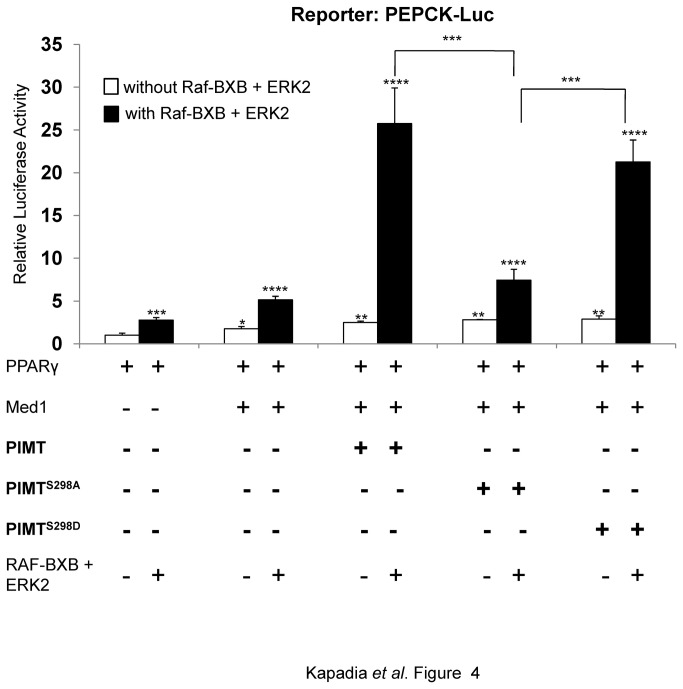
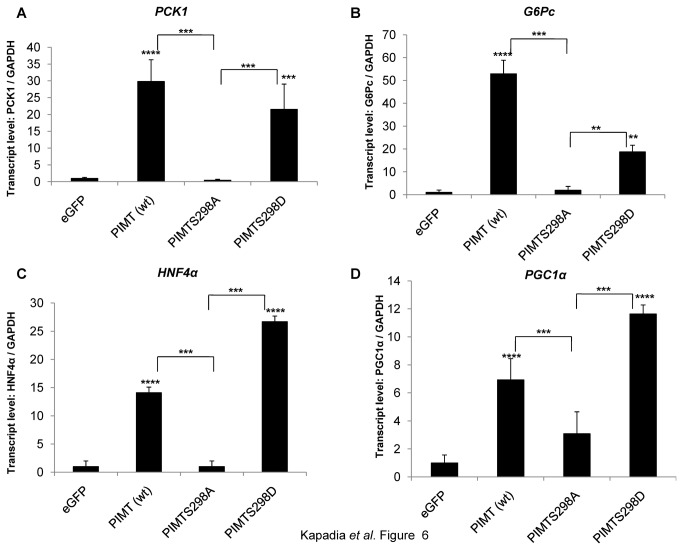
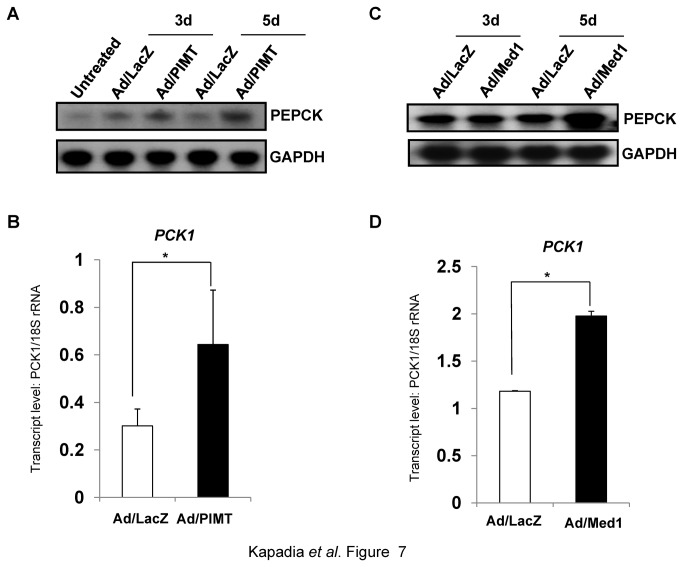
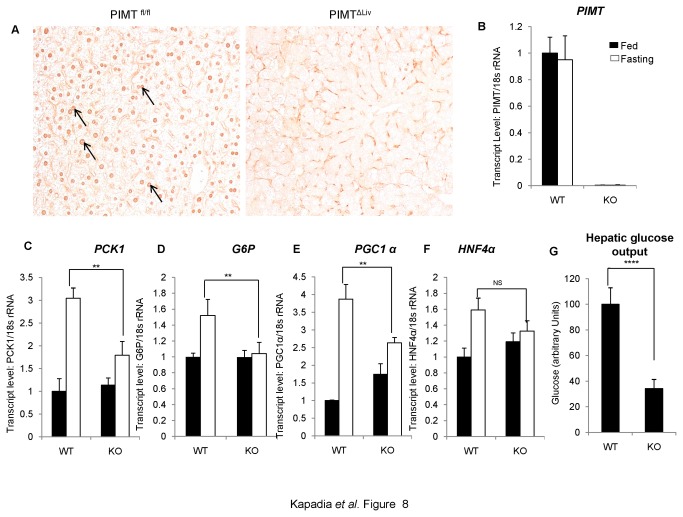
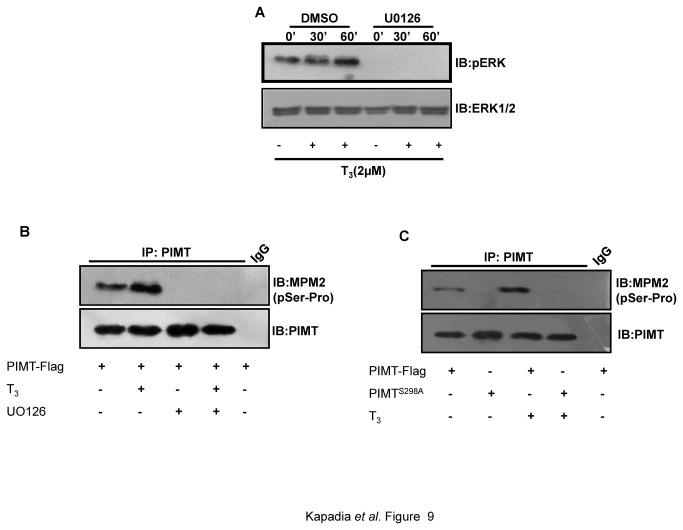
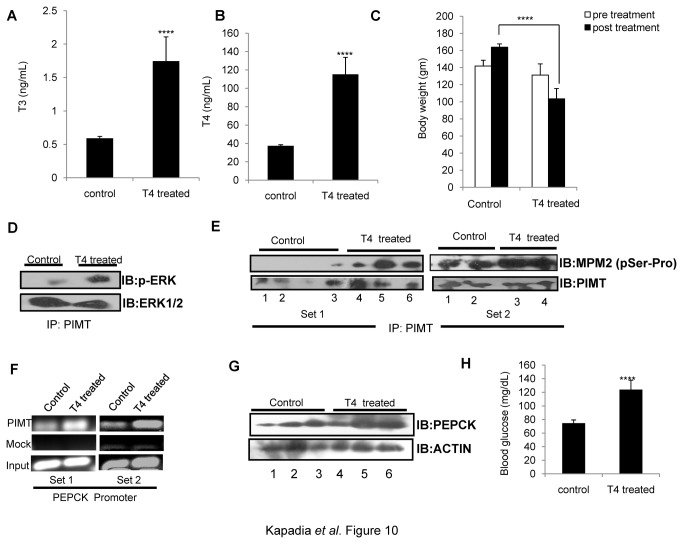
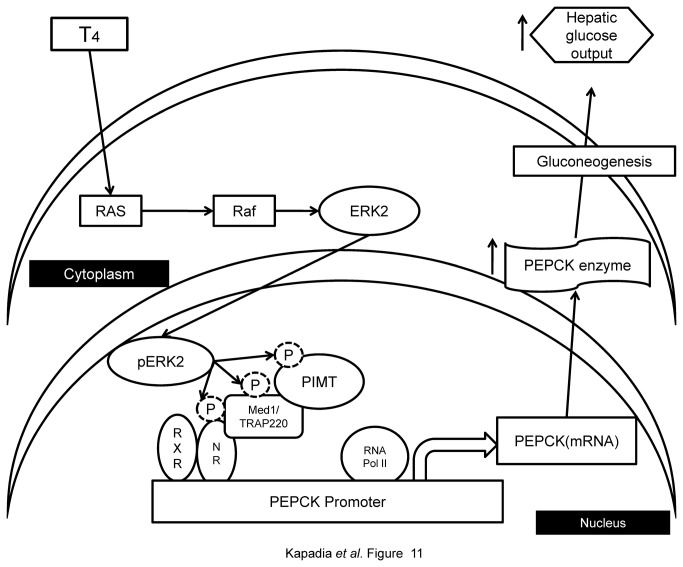
PRIP-Interacting protein with methyl transferase domain (PIMT) serves as a molecular bridge between CREB-binding protein (CBP)/ E1A binding protein p300 (Ep300) -anchored histone acetyl transferase and the Mediator complex sub-unit1 (Med1) and modulates nuclear receptor transcription. Here, we report that ERK2 phosphorylates PIMT at Ser(298) and enhances its ability to activate PEPCK promoter. We observed that PIMT is recruited to PEPCK promoter and adenoviral-mediated over-expression of PIMT in rat primary hepatocytes up-regulated expression of gluconeogenic genes including PEPCK. Reporter experiments with phosphomimetic PIMT mutant (PIMT(S298D)) suggested that conformational change may play an important role in PIMT-dependent PEPCK promoter activity. Overexpression of PIMT and Med1 together augmented hepatic glucose output in an additive manner. Importantly, expression of gluconeogenic genes and hepatic glucose output were suppressed in isolated liver specific PIMT knockout mouse hepatocytes. Furthermore, consistent with reporter experiments, PIMT(S298D) but not PIMT(S298A) augmented hepatic glucose output via up-regulating the expression of gluconeogenic genes. Pharmacological blockade of MAPK/ERK pathway using U0126, abolished PIMT/Med1-dependent gluconeogenic program leading to reduced hepatic glucose output. Further, systemic administration of T4 hormone to rats activated ERK1/2 resulting in enhanced PIMT ser(298) phosphorylation. Phosphorylation of PIMT led to its increased binding to the PEPCK promoter, increased PEPCK expression and induction of gluconeogenesis in liver. Thus, ERK2-mediated phosphorylation of PIMT at Ser(298) is essential in hepatic gluconeogenesis, demonstrating an important role of PIMT in the pathogenesis of hyperglycemia.
Conflict of interest statement
Figures

Similar articles
-
Co-activator binding protein PIMT mediates TNF-α induced insulin resistance in skeletal muscle via the transcriptional down-regulation of MEF2A and GLUT4.Sci Rep. 2015 Oct 15;5:15197. doi: 10.1038/srep15197. Sci Rep. 2015. PMID: 26468734 Free PMC article.
-
Regulation of hepatic gluconeogenesis by nuclear factor Y transcription factor in mice.J Biol Chem. 2018 May 18;293(20):7894-7904. doi: 10.1074/jbc.RA117.000508. Epub 2018 Mar 12. J Biol Chem. 2018. PMID: 29530977 Free PMC article.
-
Interaction of PIMT with transcriptional coactivators CBP, p300, and PBP differential role in transcriptional regulation.J Biol Chem. 2002 May 31;277(22):20011-9. doi: 10.1074/jbc.M201739200. Epub 2002 Mar 23. J Biol Chem. 2002. PMID: 11912212
-
AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner.J Biol Chem. 2010 Oct 15;285(42):32182-91. doi: 10.1074/jbc.M110.134890. Epub 2010 Aug 5. J Biol Chem. 2010. PMID: 20688914 Free PMC article.
-
PIMT/TGS1: An evolving metabolic molecular switch with conserved methyl transferase activity.Drug Discov Today. 2022 Aug;27(8):2386-2393. doi: 10.1016/j.drudis.2022.04.018. Epub 2022 Apr 21. Drug Discov Today. 2022. PMID: 35462043 Review.
Cited by
-
Novel eIF4A1 inhibitors with anti-tumor activity in lymphoma.Mol Med. 2022 Sep 4;28(1):101. doi: 10.1186/s10020-022-00534-0. Mol Med. 2022. PMID: 36058921 Free PMC article.
-
The MAP3K-Coding QUI-GON JINN (QGJ) Gene Is Essential to the Formation of Unreduced Embryo Sacs in Paspalum.Front Plant Sci. 2018 Oct 24;9:1547. doi: 10.3389/fpls.2018.01547. eCollection 2018. Front Plant Sci. 2018. PMID: 30405677 Free PMC article.
-
2-[2-(4-(trifluoromethyl)phenylamino)thiazol-4-yl]acetic acid (Activator-3) is a potent activator of AMPK.Sci Rep. 2018 Jun 25;8(1):9599. doi: 10.1038/s41598-018-27974-1. Sci Rep. 2018. PMID: 29942003 Free PMC article.
-
PnTgs1-like expression during reproductive development supports a role for RNA methyltransferases in the aposporous pathway.BMC Plant Biol. 2014 Nov 18;14:297. doi: 10.1186/s12870-014-0297-0. BMC Plant Biol. 2014. PMID: 25404464 Free PMC article.
-
PCMT1 confirmed as a pan-cancer immune biomarker and a contributor to breast cancer metastasis.Am J Cancer Res. 2024 Aug 25;14(8):3711-3732. doi: 10.62347/TYLL7952. eCollection 2024. Am J Cancer Res. 2024. PMID: 39267673 Free PMC article.
References
-
- Zhu Y, Qi C, Cao WQ, Yeldandi AV, Rao MS et al. (2001) Cloning and characterization of PIMT, a protein with a methyltransferase domain, which interacts with and enhances nuclear receptor coactivator PRIP function. Proc Natl Acad Sci U S A 98: 10380-10385. doi:10.1073/pnas.181347498. PubMed: 11517327. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
