

MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction
- PMID: 24362263
- PMCID: PMC4248023
- DOI: 10.1158/2159-8290.CD-13-0235
MEK-dependent negative feedback underlies BCR-ABL-mediated oncogene addiction
Abstract
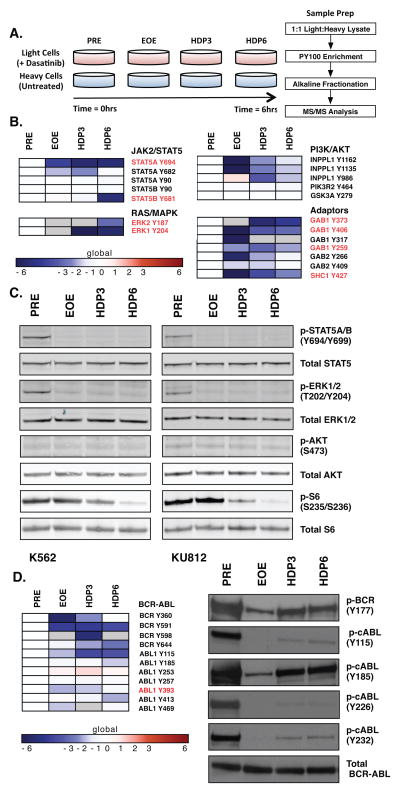
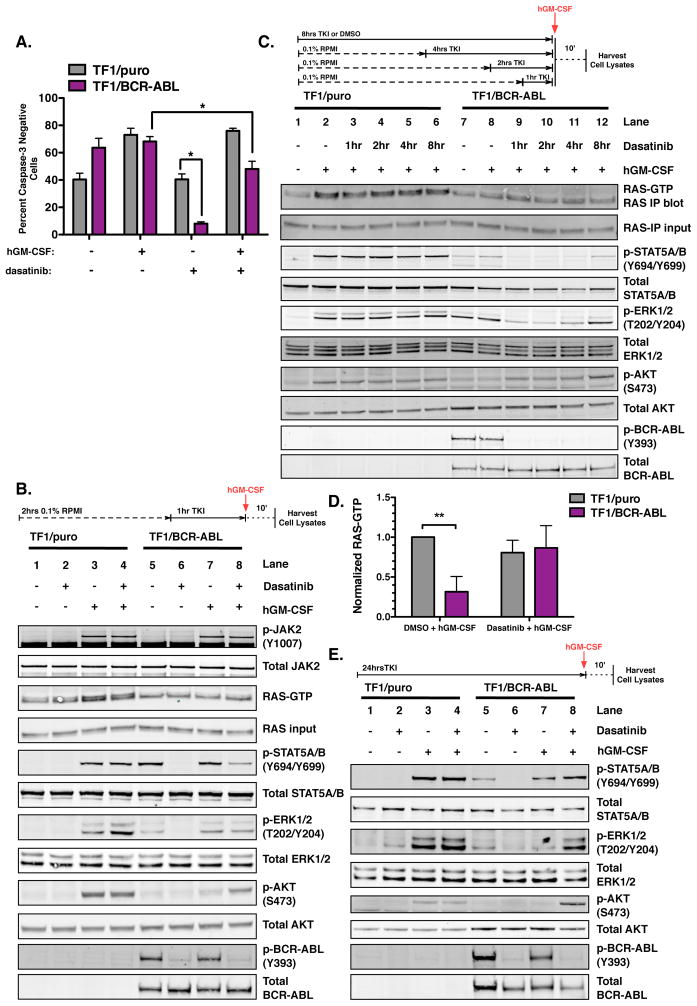
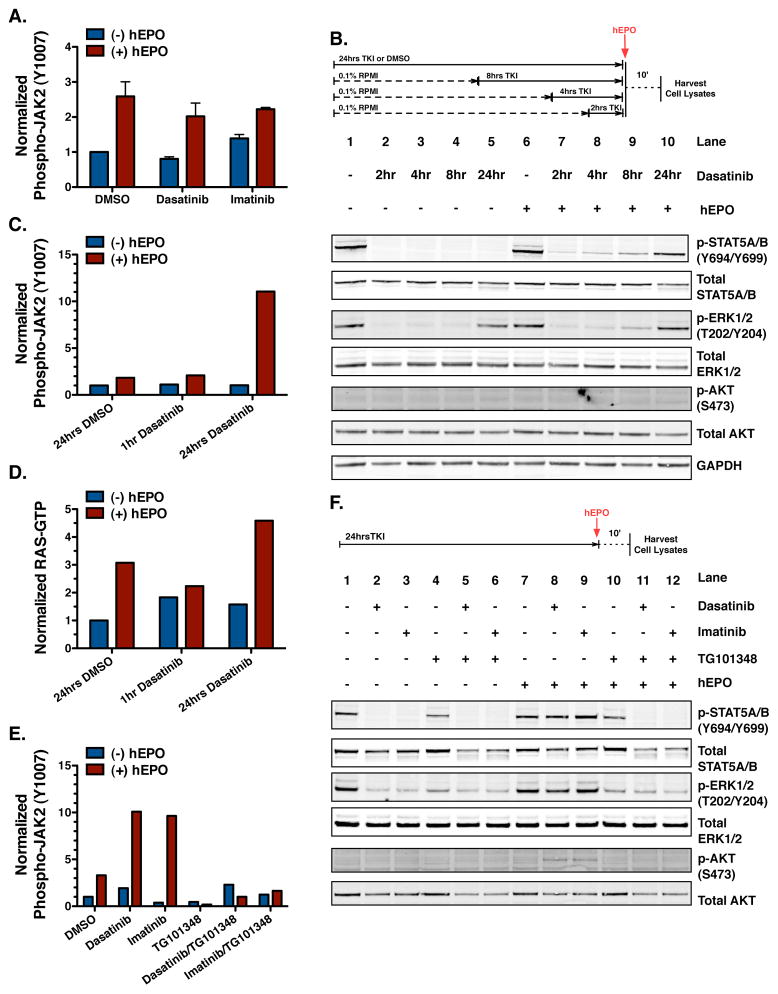
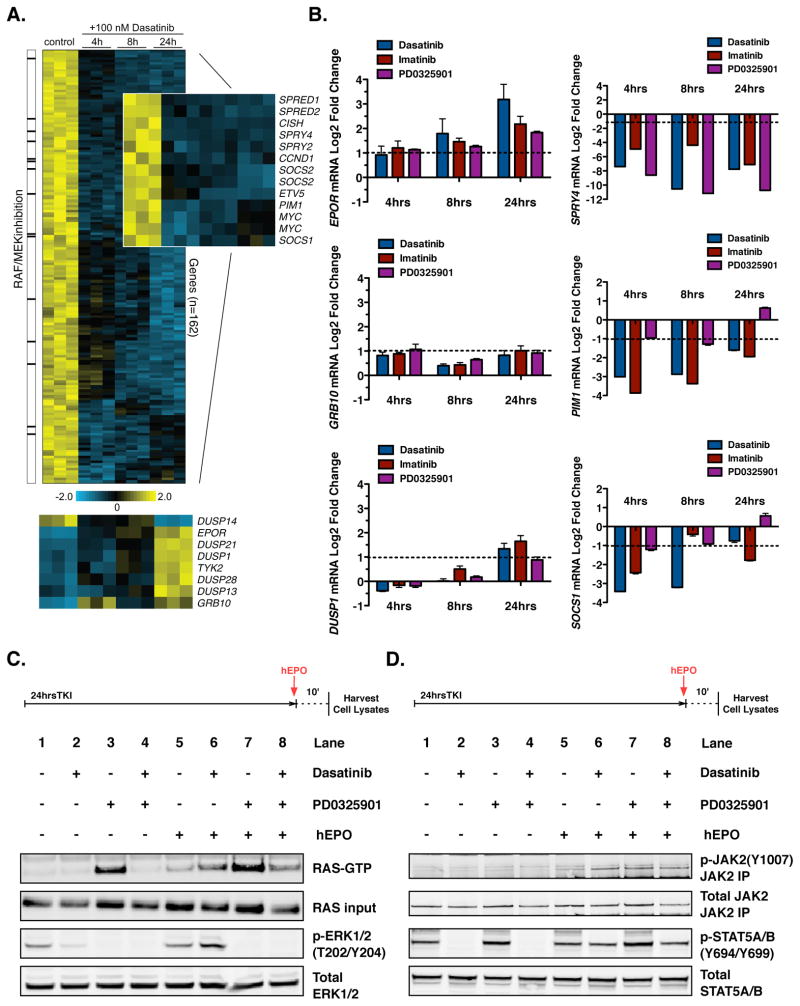
The clinical experience with BCR-ABL tyrosine kinase inhibitors (TKI) for the treatment of chronic myelogenous leukemia (CML) provides compelling evidence for oncogene addiction. Yet, the molecular basis of oncogene addiction remains elusive. Through unbiased quantitative phosphoproteomic analyses of CML cells transiently exposed to BCR-ABL TKI, we identified persistent downregulation of growth factor receptor (GF-R) signaling pathways. We then established and validated a tissue-relevant isogenic model of BCR-ABL-mediated addiction, and found evidence for myeloid GF-R signaling pathway rewiring that profoundly and persistently dampens physiologic pathway activation. We demonstrate that eventual restoration of ligand-mediated GF-R pathway activation is insufficient to fully rescue cells from a competing apoptotic fate. In contrast to previous work with BRAF(V600E) in melanoma cells, feedback inhibition following BCR-ABL TKI treatment is markedly prolonged, extending beyond the time required to initiate apoptosis. Mechanistically, BCR-ABL-mediated oncogene addiction is facilitated by persistent high levels of MAP-ERK kinase (MEK)-dependent negative feedback.
Significance: We found that BCR–ABL can confer addiction in vitro by rewiring myeloid GF-R signaling through establishment of MEK-dependent negative feedback. Our findings predict that deeper, more durable responses to targeted agents across a range of malignancies may be facilitated by maintaining negative feedback concurrently with oncoprotein inhibition.
2013 AACR
Conflict of interest statement
Conflict of Interest Disclosure Statement
N.P. Shah has received funding for the conduct of clinical research from Bristol-Myers Squibb and Ariad Pharmaceuticals. N.P. Shah has served as an ad hoc consultant for Bristol-Myers Squibb and Ariad Pharmaceuticals.
Figures
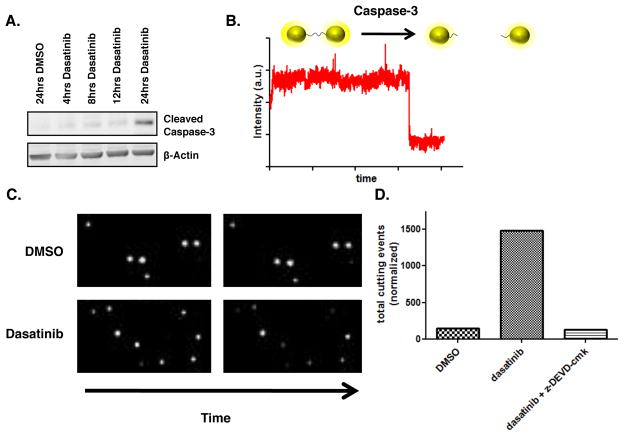
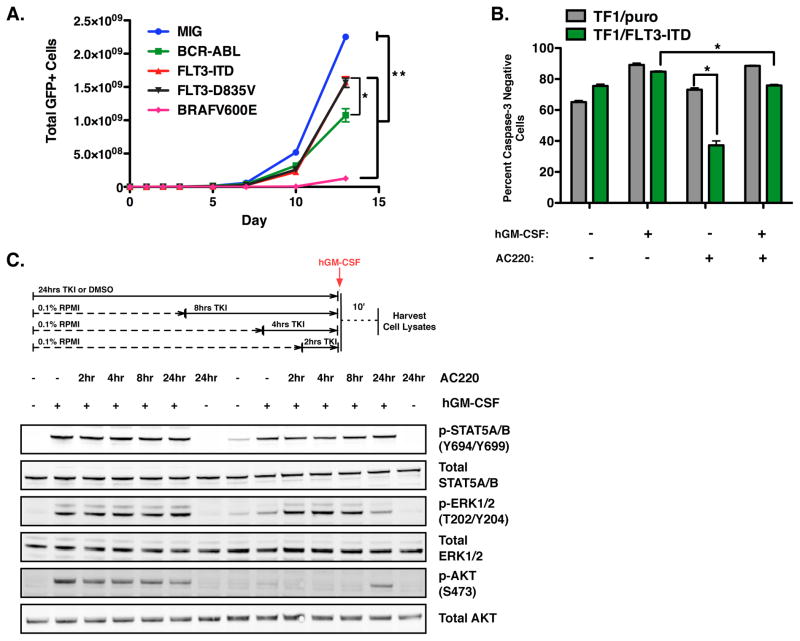
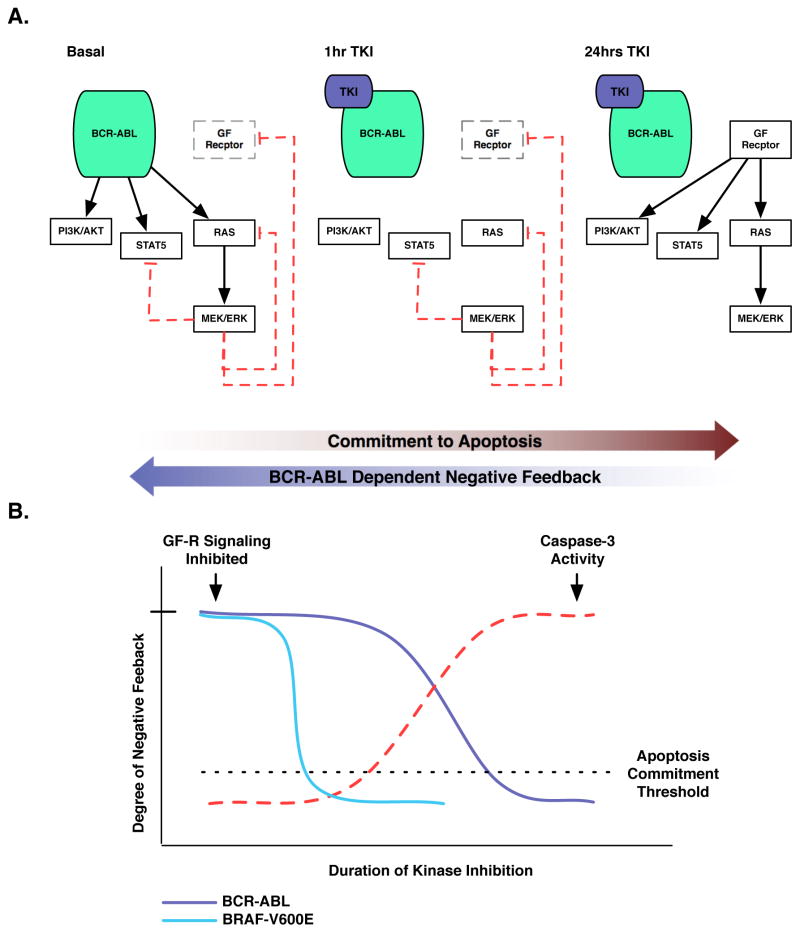
Similar articles
-
Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival.Blood. 2012 Feb 9;119(6):1501-10. doi: 10.1182/blood-2010-12-326843. Epub 2011 Dec 19. Blood. 2012. PMID: 22184410 Free PMC article.
-
Novel Mechanism by a Bis-Pyridinium Fullerene Derivative to Induce Apoptosis by Enhancing the MEK-ERK Pathway in a Reactive Oxygen Species-Independent Manner in BCR-ABL-Positive Chronic Myeloid Leukemia-Derived K562 Cells.Int J Mol Sci. 2022 Jan 11;23(2):749. doi: 10.3390/ijms23020749. Int J Mol Sci. 2022. PMID: 35054935 Free PMC article.
-
Blocking cytokine signaling along with intense Bcr-Abl kinase inhibition induces apoptosis in primary CML progenitors.Leukemia. 2010 Apr;24(4):771-8. doi: 10.1038/leu.2009.299. Epub 2010 Feb 4. Leukemia. 2010. PMID: 20130598
-
Overcoming Resistance to Targeted Therapies in Cancer.Semin Oncol. 2015 Dec;42(6):896-908. doi: 10.1053/j.seminoncol.2015.09.028. Epub 2015 Sep 25. Semin Oncol. 2015. PMID: 26615134 Review.
-
Concise review: cancer cells escape from oncogene addiction: understanding the mechanisms behind treatment failure for more effective targeting.Stem Cells. 2014 Jun;32(6):1373-9. doi: 10.1002/stem.1678. Stem Cells. 2014. PMID: 24520002 Review.
Cited by
-
The N-terminal domain of the non-receptor tyrosine kinase ABL confers protein instability and suppresses tumorigenesis.J Biol Chem. 2020 Jul 3;295(27):9069-9075. doi: 10.1074/jbc.RA120.012821. Epub 2020 May 21. J Biol Chem. 2020. PMID: 32439806 Free PMC article.
-
Knockout Serum Replacement Promotes Cell Survival by Preventing BIM from Inducing Mitochondrial Cytochrome C Release.PLoS One. 2015 Oct 16;10(10):e0140585. doi: 10.1371/journal.pone.0140585. eCollection 2015. PLoS One. 2015. PMID: 26473951 Free PMC article.
-
Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors.Hematol Oncol Clin North Am. 2017 Aug;31(4):589-612. doi: 10.1016/j.hoc.2017.04.007. Hematol Oncol Clin North Am. 2017. PMID: 28673390 Free PMC article. Review.
-
The FABD domain is critical for the oncogenicity of BCR/ABL in chronic myeloid leukaemia.Cell Commun Signal. 2024 Jun 7;22(1):314. doi: 10.1186/s12964-024-01694-8. Cell Commun Signal. 2024. PMID: 38849885 Free PMC article.
-
Pterostilbene downregulates BCR/ABL and induces apoptosis of T315I-mutated BCR/ABL-positive leukemic cells.Sci Rep. 2022 Jan 13;12(1):704. doi: 10.1038/s41598-021-04654-1. Sci Rep. 2022. PMID: 35027628 Free PMC article.
References
-
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. - PubMed
-
- Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R25-GM56847/GM/NIGMS NIH HHS/United States
- R21 EB015088/EB/NIBIB NIH HHS/United States
- R01CA128765/CA/NCI NIH HHS/United States
- 1R21EB015088/EB/NIBIB NIH HHS/United States
- T32 CA108462/CA/NCI NIH HHS/United States
- T32 GM064337/GM/NIGMS NIH HHS/United States
- R25 GM056847/GM/NIGMS NIH HHS/United States
- T32 GM007175/GM/NIGMS NIH HHS/United States
- T32CA108462-07/CA/NCI NIH HHS/United States
- P41 GM103481/GM/NIGMS NIH HHS/United States
- 8P41GM103481/GM/NIGMS NIH HHS/United States
- R01 CA128765/CA/NCI NIH HHS/United States
- P41 RR001614/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
