

Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma
- PMID: 24362818
- PMCID: PMC3907271
- DOI: 10.1038/ng.2856
Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma
Abstract
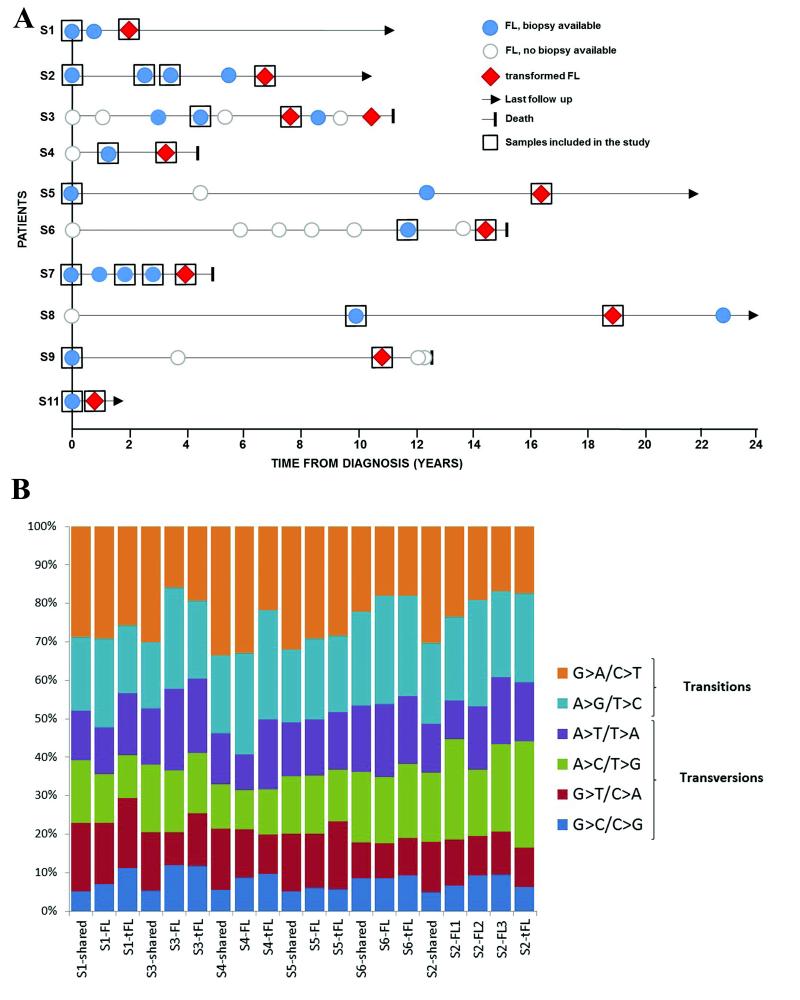
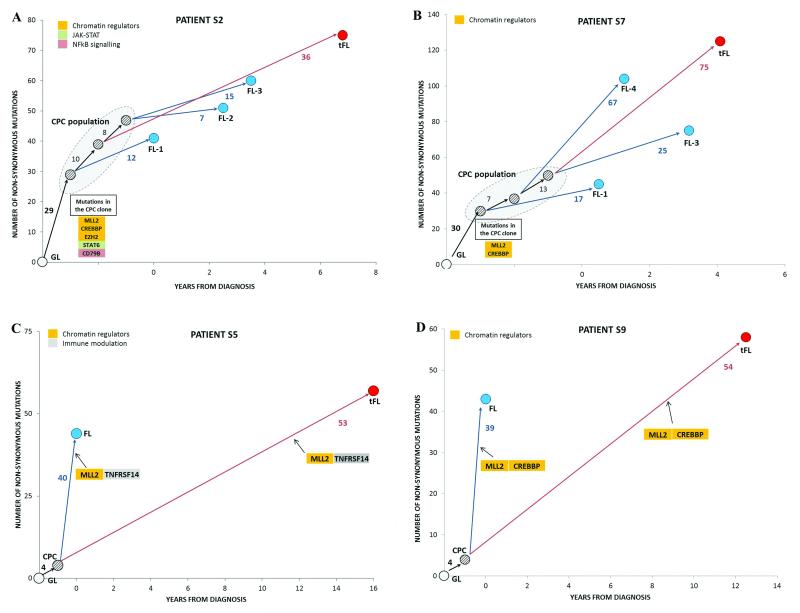
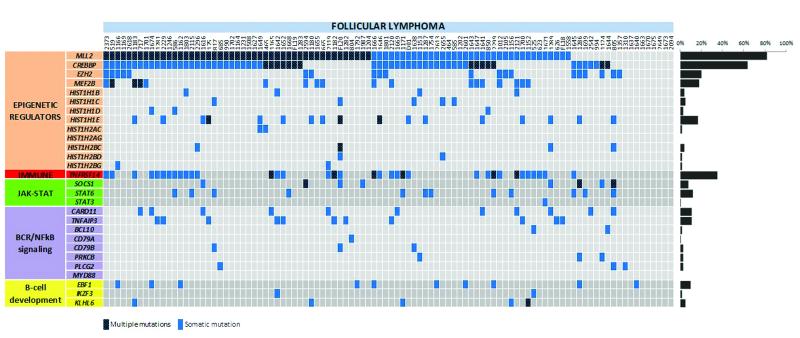
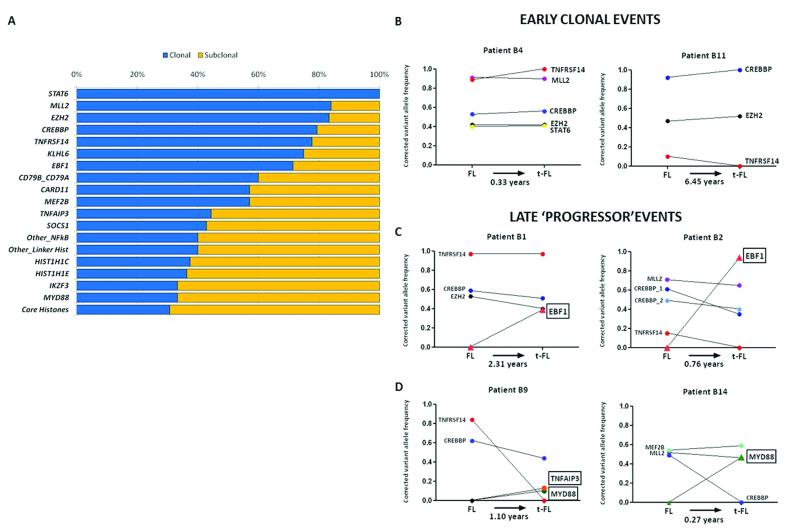
Follicular lymphoma is an incurable malignancy, with transformation to an aggressive subtype representing a critical event during disease progression. Here we performed whole-genome or whole-exome sequencing on 10 follicular lymphoma-transformed follicular lymphoma pairs followed by deep sequencing of 28 genes in an extension cohort, and we report the key events and evolutionary processes governing tumor initiation and transformation. Tumor evolution occurred through either a 'rich' or 'sparse' ancestral common progenitor clone (CPC). We identified recurrent mutations in linker histone, JAK-STAT signaling, NF-κB signaling and B cell developmental genes. Longitudinal analyses identified early driver mutations in chromatin regulator genes (CREBBP, EZH2 and KMT2D (MLL2)), whereas mutations in EBF1 and regulators of NF-κB signaling (MYD88 and TNFAIP3) were gained at transformation. Collectively, this study provides new insights into the genetic basis of follicular lymphoma and the clonal dynamics of transformation and suggests that personalizing therapies to target key genetic alterations in the CPC represents an attractive therapeutic strategy.
Figures

References
-
- Swenson WT, et al. Improved survival of follicular lymphoma patients in the United States. J Clin Oncol. 2005;23:5019–26. - PubMed
-
- Al-Tourah AJ, et al. Population-based analysis of incidence and outcome of transformed non-Hodgkin’s lymphoma. J Clin Oncol. 2008;26:5165–9. - PubMed
-
- Montoto S, Fitzgibbon J. Transformation of indolent B-cell lymphomas. J Clin Oncol. 2011;29:1827–34. - PubMed
-
- Montoto S, et al. Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J Clin Oncol. 2007;25:2426–33. - PubMed
-
- Carlotti E, et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma may occur by divergent evolution from a common progenitor cell or by direct evolution from the follicular lymphoma clone. Blood. 2009;113:3553–7. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
