

Imprinted expression of UBE3A in non-neuronal cells from a Prader-Willi syndrome patient with an atypical deletion
- PMID: 24363065
- PMCID: PMC3976333
- DOI: 10.1093/hmg/ddt628
Imprinted expression of UBE3A in non-neuronal cells from a Prader-Willi syndrome patient with an atypical deletion
Abstract
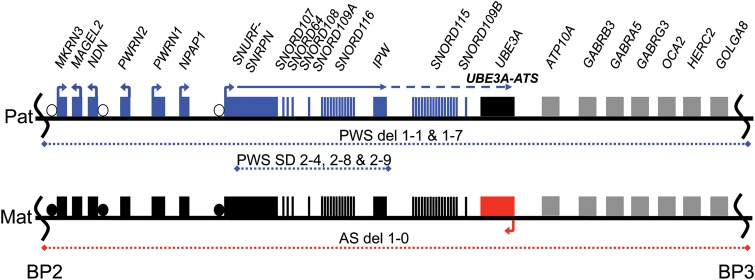
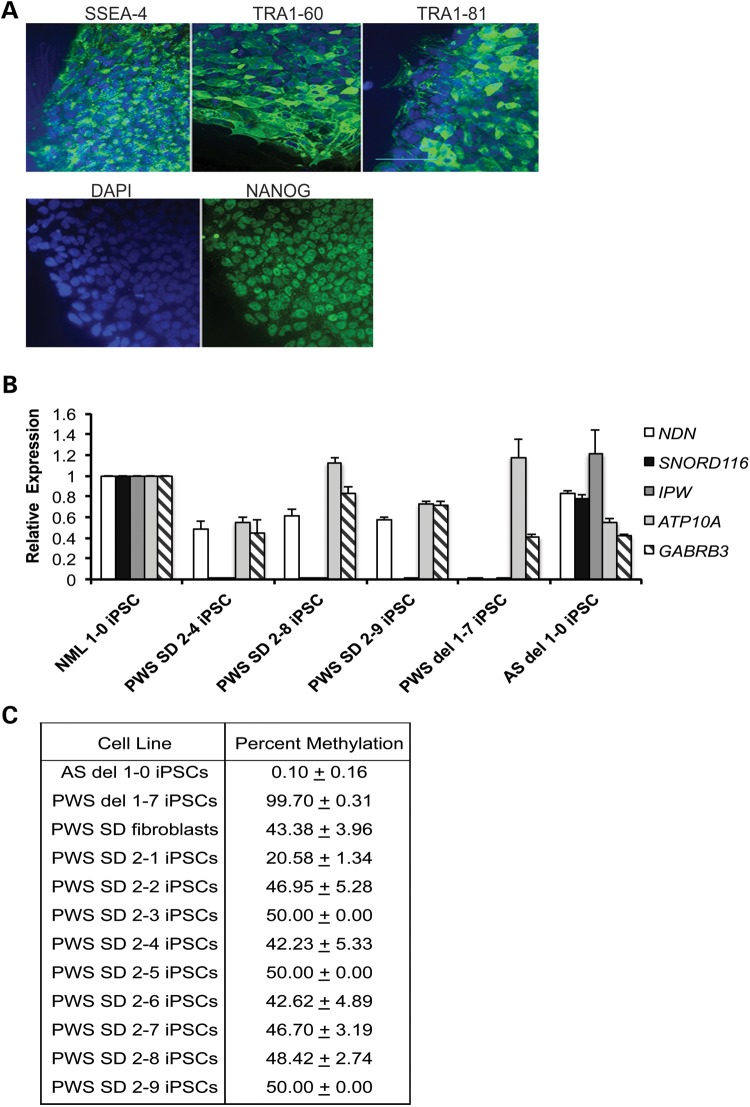
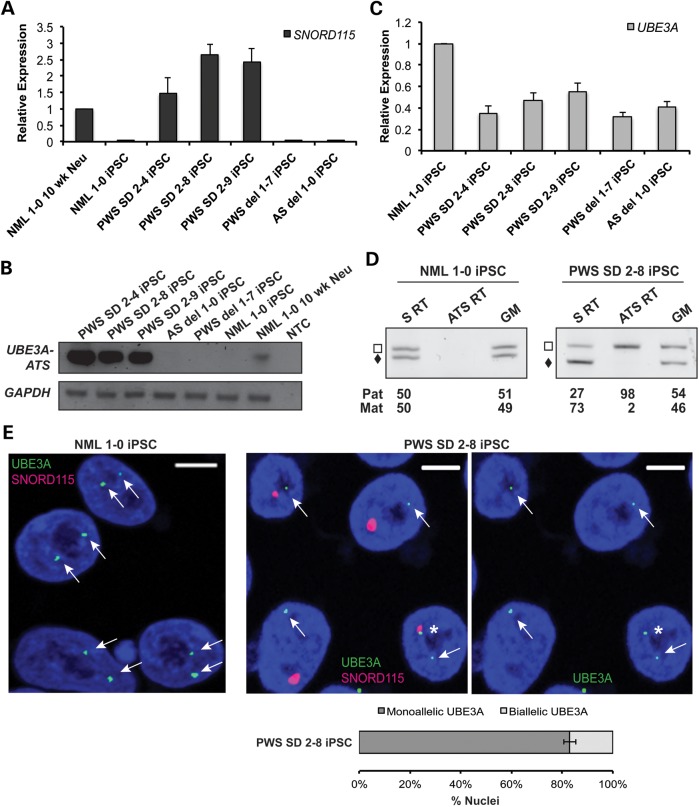
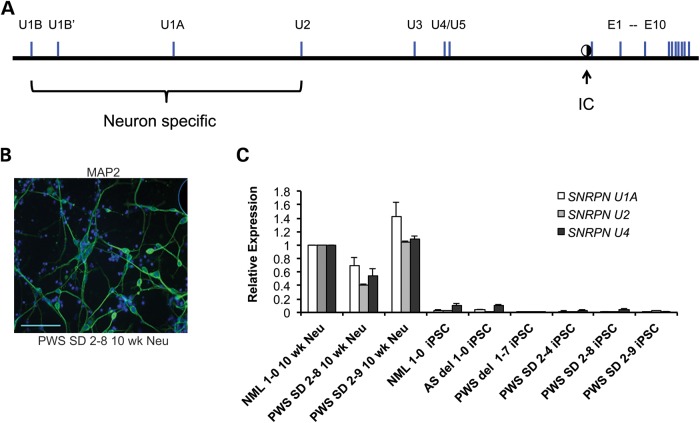
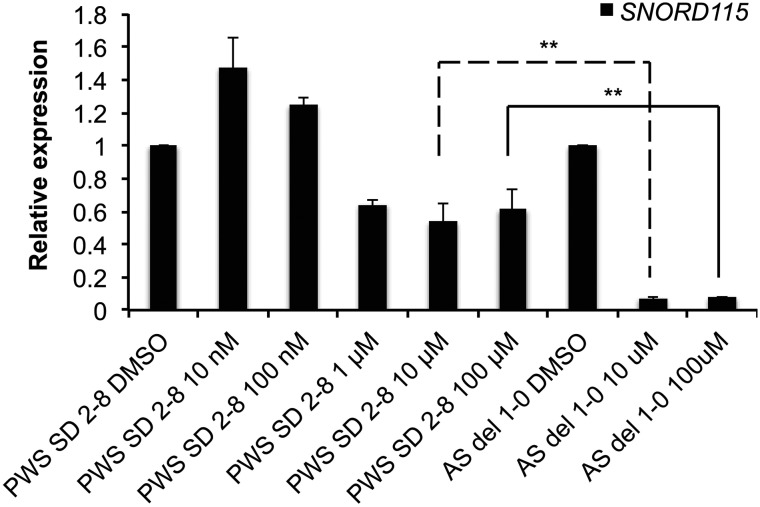
Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are two neurodevelopmental disorders most often caused by deletions of the same region of paternally inherited and maternally inherited human chromosome 15q, respectively. AS is a single gene disorder, caused by the loss of function of the ubiquitin ligase E3A (UBE3A) gene, while PWS is still considered a contiguous gene disorder. Rare individuals with PWS who carry atypical microdeletions on chromosome 15q have narrowed the critical region for this disorder to a 108 kb region that includes the SNORD116 snoRNA cluster and the Imprinted in Prader-Willi (IPW) non-coding RNA. Here we report the derivation of induced pluripotent stem cells (iPSCs) from a PWS patient with an atypical microdeletion that spans the PWS critical region. We show that these iPSCs express brain-specific portions of the transcripts driven by the PWS imprinting center, including the UBE3A antisense transcript (UBE3A-ATS). Furthermore, UBE3A expression is imprinted in most of these iPSCs. These data suggest that UBE3A imprinting in neurons only requires UBE3A-ATS expression, and no other neuron-specific factors. These data also suggest that a boundary element lying within the PWS critical region prevents UBE3A-ATS expression in non-neural tissues.
Figures
Similar articles
-
R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation.Proc Natl Acad Sci U S A. 2013 Aug 20;110(34):13938-43. doi: 10.1073/pnas.1305426110. Epub 2013 Aug 5. Proc Natl Acad Sci U S A. 2013. PMID: 23918391 Free PMC article.
-
Differential regulation of non-protein coding RNAs from Prader-Willi Syndrome locus.Sci Rep. 2014 Sep 23;4:6445. doi: 10.1038/srep06445. Sci Rep. 2014. PMID: 25246219 Free PMC article.
-
Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes.Proc Natl Acad Sci U S A. 2010 Oct 12;107(41):17668-73. doi: 10.1073/pnas.1004487107. Epub 2010 Sep 27. Proc Natl Acad Sci U S A. 2010. PMID: 20876107 Free PMC article.
-
Prader-Willi syndrome and Angelman syndrome.Am J Med Genet C Semin Med Genet. 2010 Aug 15;154C(3):365-76. doi: 10.1002/ajmg.c.30273. Am J Med Genet C Semin Med Genet. 2010. PMID: 20803659 Review.
-
Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes.Mol Hum Reprod. 1997 Apr;3(4):321-32. doi: 10.1093/molehr/3.4.321. Mol Hum Reprod. 1997. PMID: 9237260 Review.
Cited by
-
GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility.Neuron. 2016 Apr 6;90(1):56-69. doi: 10.1016/j.neuron.2016.02.040. Epub 2016 Mar 24. Neuron. 2016. PMID: 27021170 Free PMC article.
-
Integration of CTCF loops, methylome, and transcriptome in differentiating LUHMES as a model for imprinting dynamics of the 15q11-q13 locus in human neurons.Hum Mol Genet. 2024 Sep 19;33(19):1711-1725. doi: 10.1093/hmg/ddae111. Hum Mol Genet. 2024. PMID: 39045627 Free PMC article.
-
Genomic imprinting does not reduce the dosage of UBE3A in neurons.Epigenetics Chromatin. 2017 May 15;10:27. doi: 10.1186/s13072-017-0134-4. eCollection 2017. Epigenetics Chromatin. 2017. PMID: 28515788 Free PMC article.
-
Transflammation: Innate immune signaling in nuclear reprogramming.Adv Drug Deliv Rev. 2017 Oct 1;120:133-141. doi: 10.1016/j.addr.2017.09.010. Epub 2017 Sep 13. Adv Drug Deliv Rev. 2017. PMID: 28916494 Free PMC article. Review.
-
Studying Abnormal Chromosomal Diseases Using Patient-Derived Induced Pluripotent Stem Cells.Front Cell Neurosci. 2020 Aug 13;14:224. doi: 10.3389/fncel.2020.00224. eCollection 2020. Front Cell Neurosci. 2020. PMID: 32922264 Free PMC article.
References
-
- Cassidy S.B., Dykens E., Williams C.A. Prader–Willi and Angelman syndromes: sister imprinted disorders. Am. J. Med. Genet. 2000;97:136–146. - PubMed
-
- Williams C.A., Beaudet A.L., Clayton-Smith J., Knoll J.H., Kyllerman M., Laan L.A., Magenis R.E., Moncla A., Schinzel A.A., Summers J.A., et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am. J. Med. Genet. A. 2006;140:413–418. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
