

Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy
- PMID: 24366076
- PMCID: PMC3929844
- DOI: 10.1161/HYPERTENSIONAHA.113.02219
Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy
Abstract
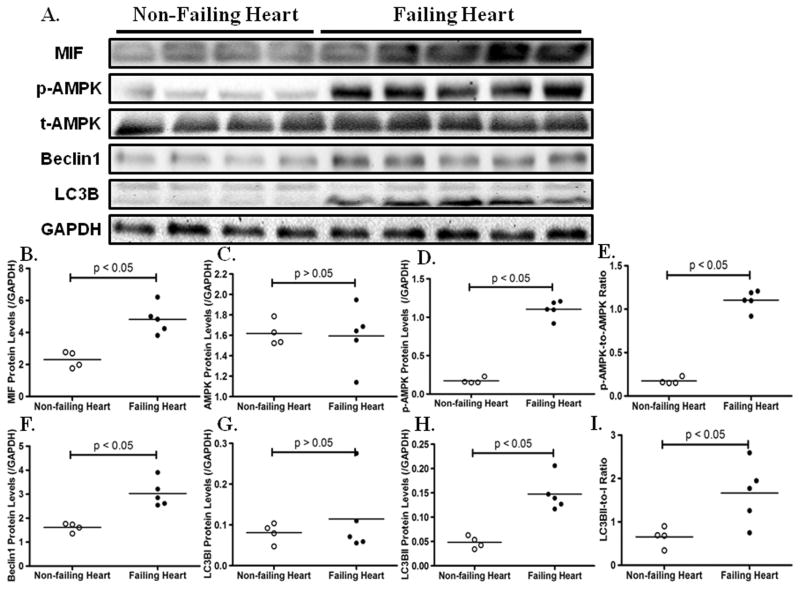
The proinflammatory cytokine macrophage migration inhibitory factor (MIF) has been shown to be cardioprotective under various pathological conditions. However, the underlying mechanisms still remain elusive. In this study, we revealed that MIF deficiency overtly exacerbated abdominal aorta constriction-induced cardiac hypertrophy and contractile anomalies. MIF deficiency interrupted myocardial autophagy in hypertrophied hearts. Rapamycin administration mitigated the exacerbated hypertrophic responses in MIF(-/-) mice. Using the phenylephrine-induced hypertrophy in vitro model in H9C2 myoblasts, we confirmed that MIF governed the activation of AMP-activated protein kinase-mammalian target of rapamycin-autophagy cascade. Confocal microscopic examination demonstrated that MIF depletion prevented phenylephrine-induced mitophagy in H9C2 myoblasts. Myocardial Parkin, an E3 ubiquitin ligase and a marker for mitophagy, was significantly upregulated after sustained pressure overload, the effect of which was prevented by MIF knockout. Furthermore, our data exhibited that levels of MIF, AMP-activated protein kinase activation, and autophagy were elevated concurrently in human failing hearts. These data indicate that endogenous MIF regulates the mammalian target of rapamycin signaling to activate autophagy to preserve cardiac geometry and protect against hypertrophic responses.
Keywords: autophagy; cardiac hypertrophy; macrophage migration inhibitory factor; mammalian target of rapamycin; rapamycin.
Conflict of interest statement
None.
Figures






References
-
- Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199–204. - PubMed
-
- Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–1891. - PubMed
-
- Brown JH, Del Re DP, Sussman MA. The rac and rho hall of fame: A decade of hypertrophic signaling hits. Circ Res. 2006;98:730–742. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
