

A disorder-induced domino-like destabilization mechanism governs the folding and functional dynamics of the repeat protein IκBα
- PMID: 24367251
- PMCID: PMC3868533
- DOI: 10.1371/journal.pcbi.1003403
A disorder-induced domino-like destabilization mechanism governs the folding and functional dynamics of the repeat protein IκBα
Abstract
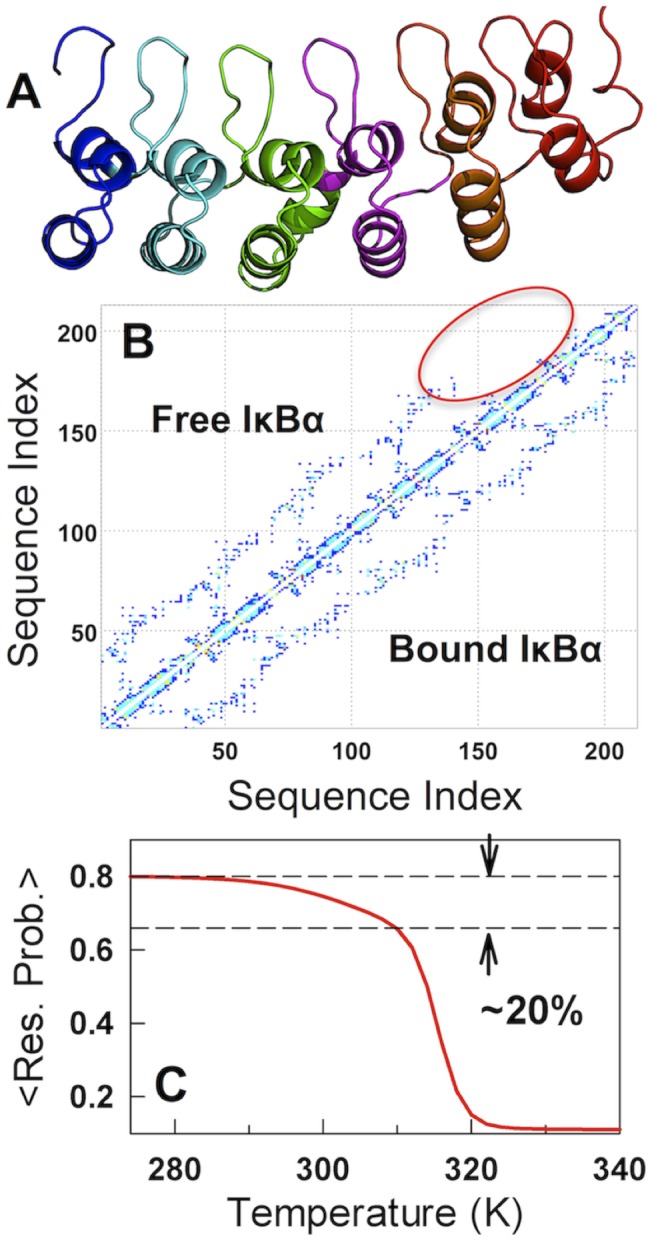
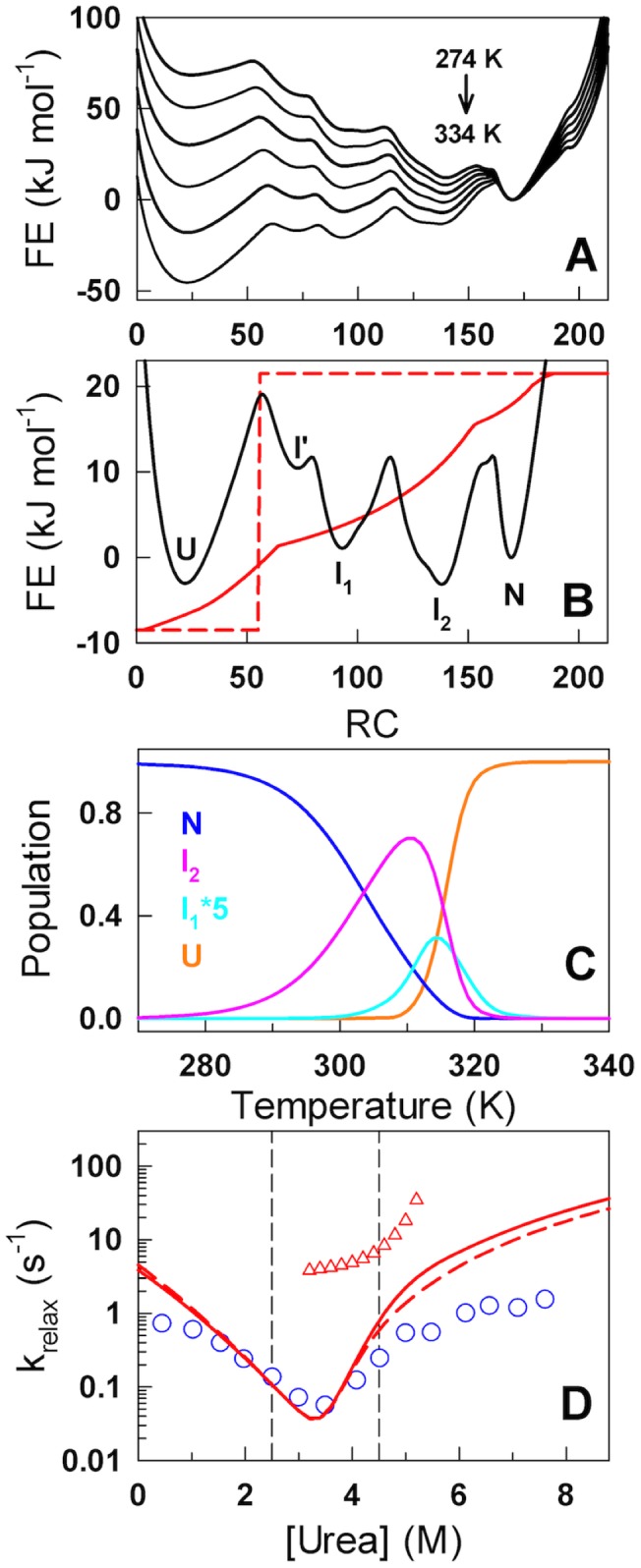
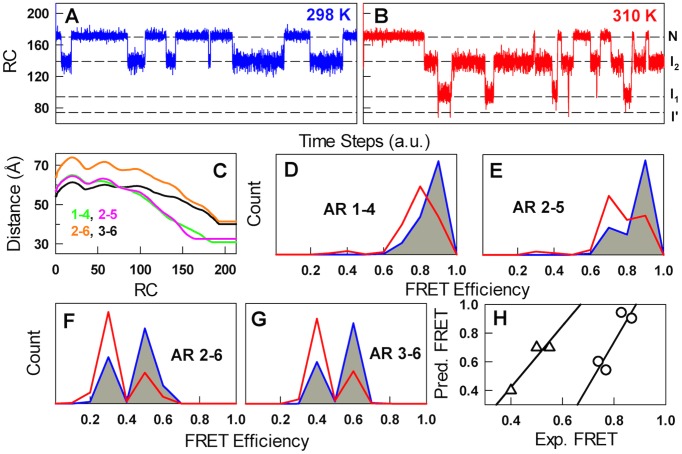
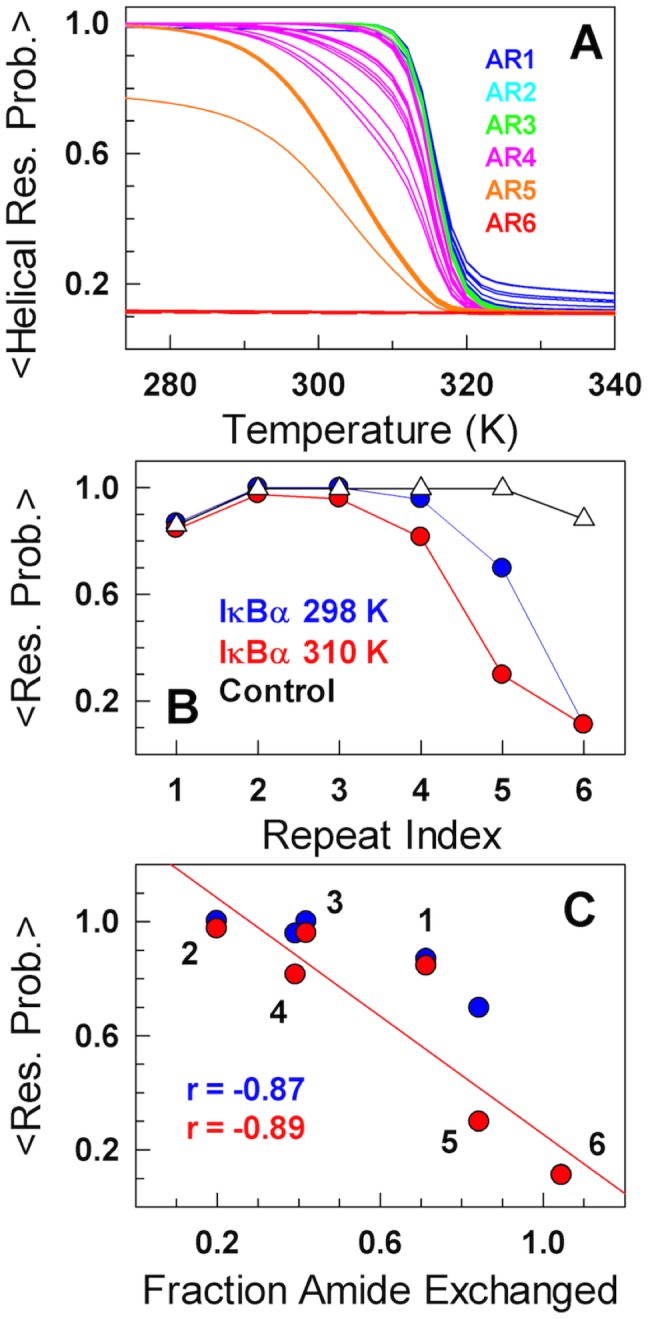
The stability of the repeat protein IκBα, a transcriptional inhibitor in mammalian cells, is critical in the functioning of the NF-κB signaling module implicated in an array of cellular processes, including cell growth, disease, immunity and apoptosis. Structurally, IκBα is complex, with both ordered and disordered regions, thus posing a challenge to the available computational protocols to model its conformational behavior. Here, we introduce a simple procedure to model disorder in systems that undergo binding-induced folding that involves modulation of the contact map guided by equilibrium experimental observables in combination with an Ising-like Wako-Saitô-Muñoz-Eaton model. This one-step procedure alone is able to reproduce a variety of experimental observables, including ensemble thermodynamics (scanning calorimetry, pre-transitions, m-values) and kinetics (roll-over in chevron plot, intermediates and their identity), and is consistent with hydrogen-deuterium exchange measurements. We further capture the intricate distance-dynamics between the domains as measured by single-molecule FRET by combining the model predictions with simple polymer physics arguments. Our results reveal a unique mechanism at work in IκBα folding, wherein disorder in one domain initiates a domino-like effect partially destabilizing neighboring domains, thus highlighting the effect of symmetry-breaking at the level of primary sequences. The offshoot is a multi-state and a dynamic conformational landscape that is populated by increasingly partially folded ensembles upon destabilization. Our results provide, in a straightforward fashion, a rationale to the promiscuous binding and short intracellular half-life of IκBα evolutionarily engineered into it through repeats with variable stabilities and expand the functional repertoire of disordered regions in proteins.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
The Wako-Saitô-Muñoz-Eaton Model for Predicting Protein Folding and Dynamics.Molecules. 2022 Jul 12;27(14):4460. doi: 10.3390/molecules27144460. Molecules. 2022. PMID: 35889332 Free PMC article. Review.
-
Stabilizing IkappaBalpha by "consensus" design.J Mol Biol. 2007 Jan 26;365(4):1201-16. doi: 10.1016/j.jmb.2006.11.044. Epub 2006 Nov 15. J Mol Biol. 2007. PMID: 17174335 Free PMC article.
-
Detection of a ternary complex of NF-kappaB and IkappaBalpha with DNA provides insights into how IkappaBalpha removes NF-kappaB from transcription sites.Proc Natl Acad Sci U S A. 2011 Jan 25;108(4):1367-72. doi: 10.1073/pnas.1014323108. Epub 2011 Jan 10. Proc Natl Acad Sci U S A. 2011. PMID: 21220295 Free PMC article.
-
Visualization of the nanospring dynamics of the IkappaBalpha ankyrin repeat domain in real time.Proc Natl Acad Sci U S A. 2011 Jun 21;108(25):10178-83. doi: 10.1073/pnas.1102226108. Epub 2011 May 31. Proc Natl Acad Sci U S A. 2011. PMID: 21628581 Free PMC article.
-
Role of disorder in IκB-NFκB interaction.IUBMB Life. 2012 Jun;64(6):499-505. doi: 10.1002/iub.1044. Epub 2012 May 9. IUBMB Life. 2012. PMID: 22573609 Free PMC article. Review.
Cited by
-
Phosphorylation of Thr9 Affects the Folding Landscape of the N-Terminal Segment of Human AGT Enhancing Protein Aggregation of Disease-Causing Mutants.Molecules. 2022 Dec 10;27(24):8762. doi: 10.3390/molecules27248762. Molecules. 2022. PMID: 36557898 Free PMC article.
-
Protein plasticity driven by disorder and collapse governs the heterogeneous binding of CytR to DNA.Nucleic Acids Res. 2018 May 4;46(8):4044-4053. doi: 10.1093/nar/gky176. Nucleic Acids Res. 2018. PMID: 29538715 Free PMC article.
-
Cooperativity and modularity in protein folding.Biophys Physicobiol. 2016 Nov 18;13:281-293. doi: 10.2142/biophysico.13.0_281. eCollection 2016. Biophys Physicobiol. 2016. PMID: 28409080 Free PMC article. Review.
-
Role of the NFκB-signaling pathway in cancer.Onco Targets Ther. 2018 Apr 11;11:2063-2073. doi: 10.2147/OTT.S161109. eCollection 2018. Onco Targets Ther. 2018. PMID: 29695914 Free PMC article. Review.
-
The Wako-Saitô-Muñoz-Eaton Model for Predicting Protein Folding and Dynamics.Molecules. 2022 Jul 12;27(14):4460. doi: 10.3390/molecules27144460. Molecules. 2022. PMID: 35889332 Free PMC article. Review.
References
-
- Courtois G, Gilmore TD (2006) Mutations in the NF-kappa B signaling pathway: implications for human disease. Oncogene 25: 6831–6843. - PubMed
-
- Orange JS, Levy O, Geha RS (2005) Human disease resulting from gene mutations that interfere with appropriate nuclear factor-kB activation. Immun Rev 203: 21–37. - PubMed
-
- Phelps CB, Sengchanthalangsy LL, Huxford T, Ghosh G (2000) Mechanism of IκBα binding to NF-κB dimers. J Biol Chem 275: 29840–29846. - PubMed
-
- Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT (1999) Mutations in the IkBa gene in Hodgkin's disease suggest a tumour suppressor role for IkBa. Oncogene 18: 3063–3070. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
