

Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions
- PMID: 24367273
- PMCID: PMC3868540
- DOI: 10.1371/journal.pgen.1003994
Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions
Abstract
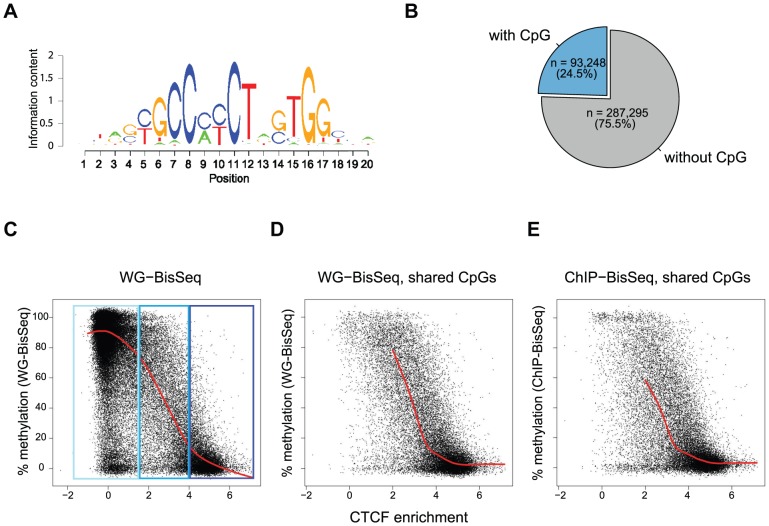
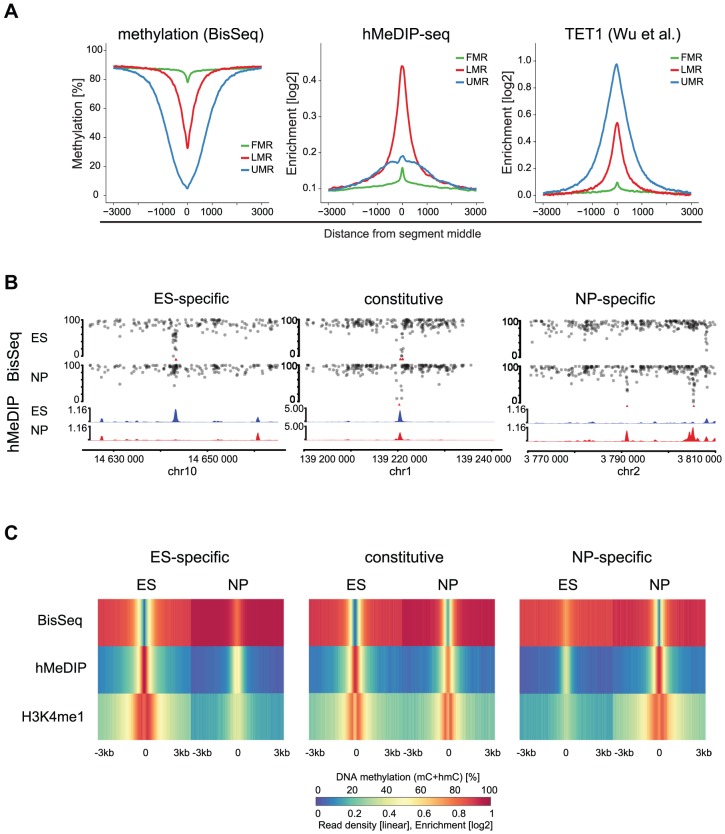
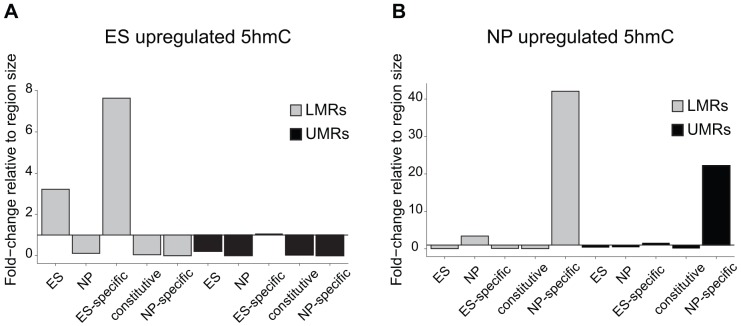
Distal regulatory elements, including enhancers, play a critical role in regulating gene activity. Transcription factor binding to these elements correlates with Low Methylated Regions (LMRs) in a process that is poorly understood. Here we ask whether and how actual occupancy of DNA-binding factors is linked to DNA methylation at the level of individual molecules. Using CTCF as an example, we observe that frequency of binding correlates with the likelihood of a demethylated state and sites of low occupancy display heterogeneous DNA methylation within the CTCF motif. In line with a dynamic model of binding and DNA methylation turnover, we find that 5-hydroxymethylcytosine (5hmC), formed as an intermediate state of active demethylation, is enriched at LMRs in stem and somatic cells. Moreover, a significant fraction of changes in 5hmC during differentiation occurs at these regions, suggesting that transcription factor activity could be a key driver for active demethylation. Since deletion of CTCF is lethal for embryonic stem cells, we used genetic deletion of REST as another DNA-binding factor implicated in LMR formation to test this hypothesis. The absence of REST leads to a decrease of hydroxymethylation and a concomitant increase of DNA methylation at its binding sites. These data support a model where DNA-binding factors can mediate turnover of DNA methylation as an integral part of maintenance and reprogramming of regulatory regions.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


References
-
- Okita K, Ichisaka T, Yamanaka S (2007) Generation of germline-competent induced pluripotent stem cells. Nature 448: 313–317. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
