

Huntington's disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart
- PMID: 24367279
- PMCID: PMC3868535
- DOI: 10.1371/journal.pgen.1004024
Huntington's disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart
Abstract
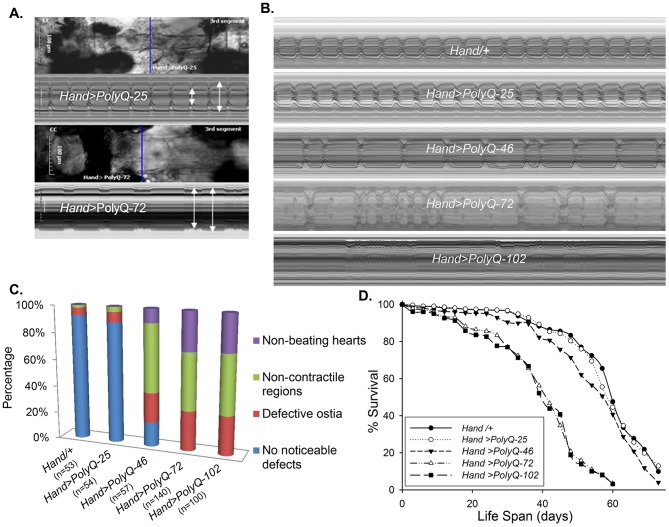
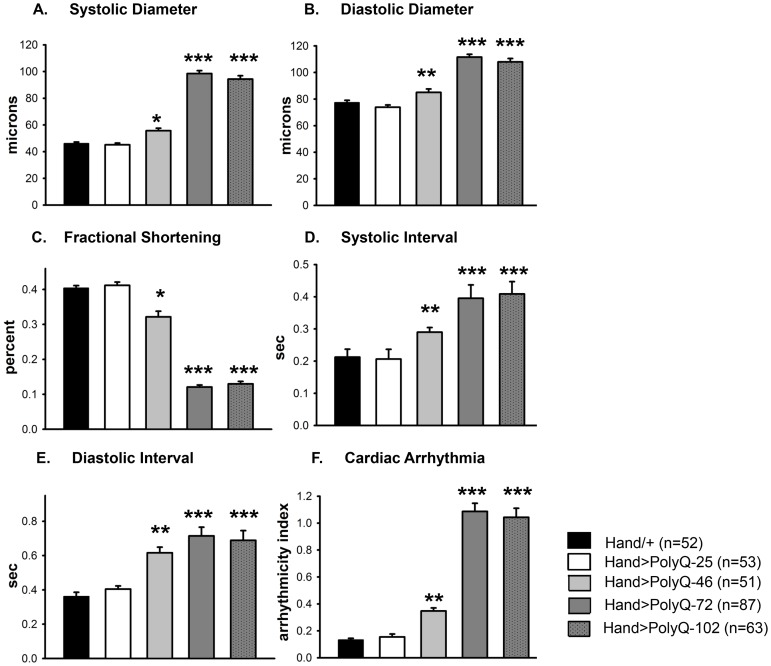
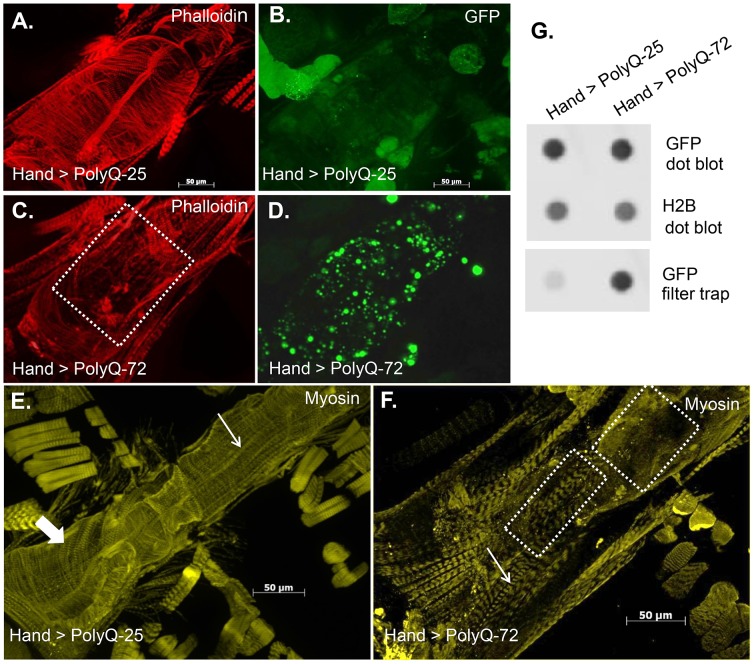
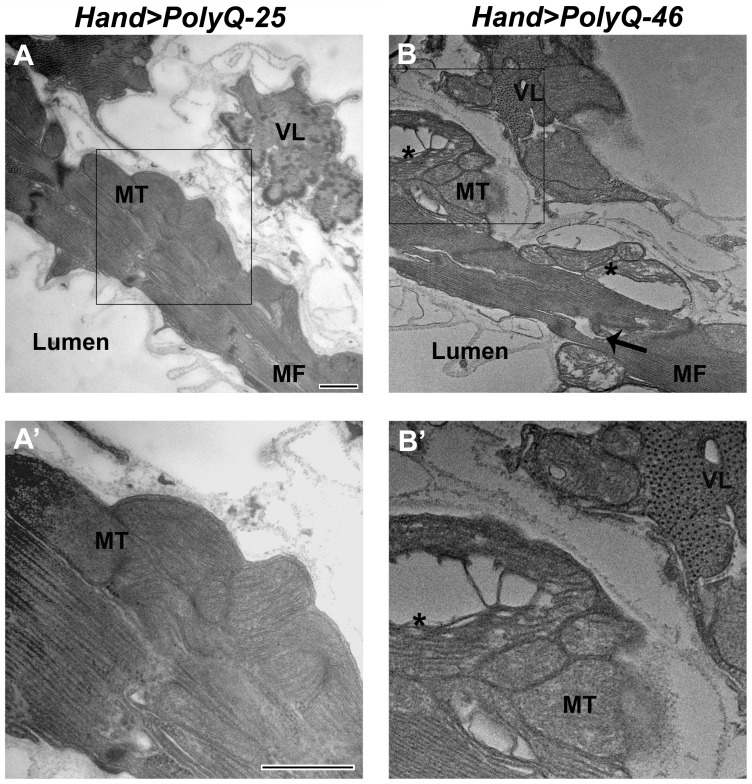
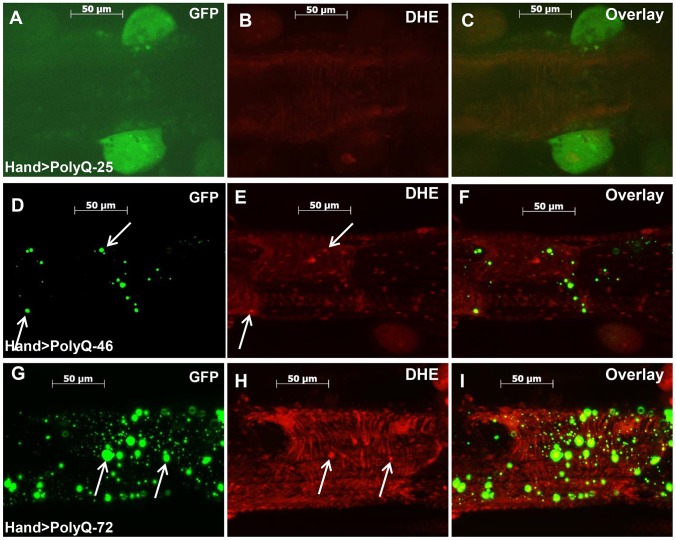
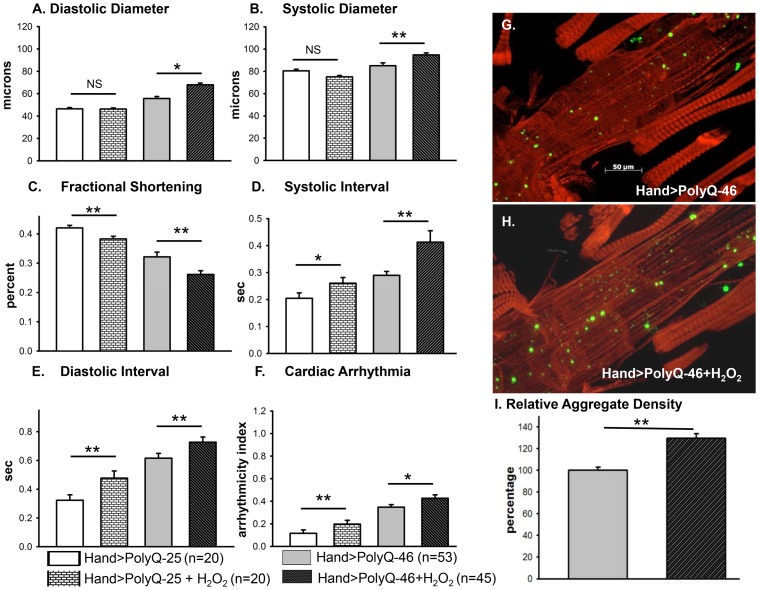
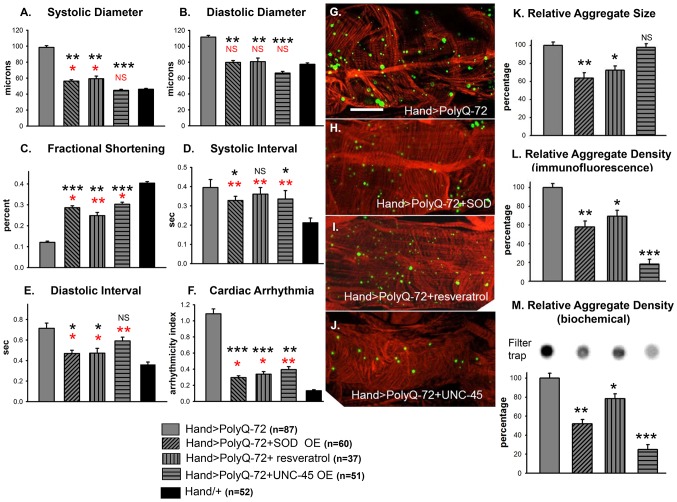
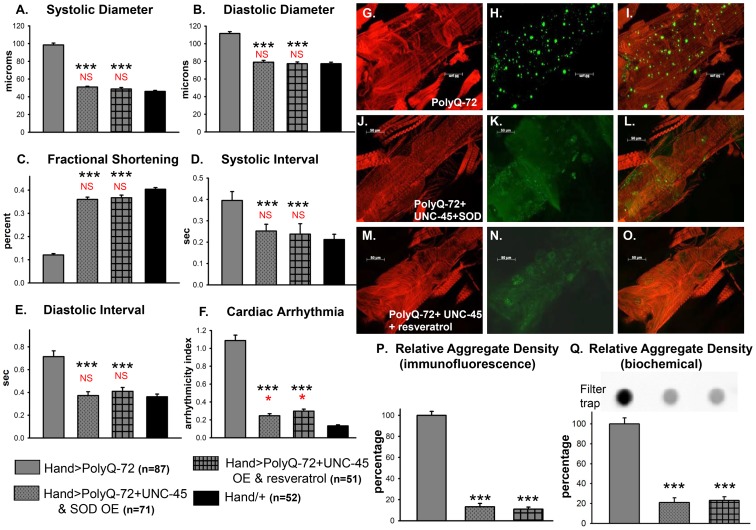
Amyloid-like inclusions have been associated with Huntington's disease (HD), which is caused by expanded polyglutamine repeats in the Huntingtin protein. HD patients exhibit a high incidence of cardiovascular events, presumably as a result of accumulation of toxic amyloid-like inclusions. We have generated a Drosophila model of cardiac amyloidosis that exhibits accumulation of PolyQ aggregates and oxidative stress in myocardial cells, upon heart-specific expression of Huntingtin protein fragments (Htt-PolyQ) with disease-causing poly-glutamine repeats (PolyQ-46, PolyQ-72, and PolyQ-102). Cardiac expression of GFP-tagged Htt-PolyQs resulted in PolyQ length-dependent functional defects that included increased incidence of arrhythmias and extreme cardiac dilation, accompanied by a significant decrease in contractility. Structural and ultrastructural analysis of the myocardial cells revealed reduced myofibrillar content, myofibrillar disorganization, mitochondrial defects and the presence of PolyQ-GFP positive aggregates. Cardiac-specific expression of disease causing Poly-Q also shortens lifespan of flies dramatically. To further confirm the involvement of oxidative stress or protein unfolding and to understand the mechanism of PolyQ induced cardiomyopathy, we co-expressed expanded PolyQ-72 with the antioxidant superoxide dismutase (SOD) or the myosin chaperone UNC-45. Co-expression of SOD suppressed PolyQ-72 induced mitochondrial defects and partially suppressed aggregation as well as myofibrillar disorganization. However, co-expression of UNC-45 dramatically suppressed PolyQ-72 induced aggregation and partially suppressed myofibrillar disorganization. Moreover, co-expression of both UNC-45 and SOD more efficiently suppressed GFP-positive aggregates, myofibrillar disorganization and physiological cardiac defects induced by PolyQ-72 than did either treatment alone. Our results demonstrate that mutant-PolyQ induces aggregates, disrupts the sarcomeric organization of contractile proteins, leads to mitochondrial dysfunction and increases oxidative stress in cardiomyocytes leading to abnormal cardiac function. We conclude that modulation of both protein unfolding and oxidative stress pathways in the Drosophila heart model can ameliorate the detrimental PolyQ effects, thus providing unique insights into the genetic mechanisms underlying amyloid-induced cardiac failure in HD patients.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Drosophila as a potential model to ameliorate mutant Huntington-mediated cardiac amyloidosis.Rare Dis. 2014 Nov 3;2(1):e968003. doi: 10.4161/2167549X.2014.968003. eCollection 2014. Rare Dis. 2014. PMID: 26942103 Free PMC article.
-
Exploding the Repeat Length Paradigm while Exploring Amyloid Toxicity in Huntington's Disease.Acc Chem Res. 2020 Oct 20;53(10):2347-2357. doi: 10.1021/acs.accounts.0c00450. Epub 2020 Sep 25. Acc Chem Res. 2020. PMID: 32975927
-
Effects of flanking sequences and cellular context on subcellular behavior and pathology of mutant HTT.Hum Mol Genet. 2020 Mar 13;29(4):674-688. doi: 10.1093/hmg/ddaa001. Hum Mol Genet. 2020. PMID: 31943010 Free PMC article.
-
Spontaneous self-assembly of pathogenic huntingtin exon 1 protein into amyloid structures.Essays Biochem. 2014;56:167-80. doi: 10.1042/bse0560167. Essays Biochem. 2014. PMID: 25131594 Review.
-
Proteostasis of Huntingtin in Health and Disease.Int J Mol Sci. 2017 Jul 19;18(7):1568. doi: 10.3390/ijms18071568. Int J Mol Sci. 2017. PMID: 28753941 Free PMC article. Review.
Cited by
-
Methods to assess Drosophila heart development, function and aging.Methods. 2014 Jun 15;68(1):265-72. doi: 10.1016/j.ymeth.2014.03.031. Epub 2014 Apr 12. Methods. 2014. PMID: 24727147 Free PMC article. Review.
-
Cardiac mTORC1 Dysregulation Impacts Stress Adaptation and Survival in Huntington's Disease.Cell Rep. 2018 Apr 24;23(4):1020-1033. doi: 10.1016/j.celrep.2018.03.117. Cell Rep. 2018. PMID: 29694882 Free PMC article.
-
UNC-45A: A potential therapeutic target for malignant tumors.Heliyon. 2024 May 14;10(10):e31276. doi: 10.1016/j.heliyon.2024.e31276. eCollection 2024 May 30. Heliyon. 2024. PMID: 38803956 Free PMC article. Review.
-
Proteostasis in cardiac health and disease.Nat Rev Cardiol. 2017 Nov;14(11):637-653. doi: 10.1038/nrcardio.2017.89. Epub 2017 Jun 29. Nat Rev Cardiol. 2017. PMID: 28660894 Review.
-
Challenging muscle homeostasis uncovers novel chaperone interactions in Caenorhabditis elegans.Front Mol Biosci. 2014 Nov 6;1:21. doi: 10.3389/fmolb.2014.00021. eCollection 2014. Front Mol Biosci. 2014. PMID: 25988162 Free PMC article.
References
-
- Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med 349: 583–596. - PubMed
-
- Muchowski PJ (2002) Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron 35: 9–12. - PubMed
-
- Mackay TF, Anholt RR (2006) Of flies and man: Drosophila as a model for human complex traits. Annu Rev Genomics Hum Genet 7: 339–367. - PubMed
-
- Rubinsztein DC, Carmichael J (2003) Huntington's disease: molecular basis of neurodegeneration. Expert Rev Mol Med 5: 1–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R37 GM032443/GM/NIGMS NIH HHS/United States
- R01HL54732/HL/NHLBI NIH HHS/United States
- R01 HL054732/HL/NHLBI NIH HHS/United States
- T34GM008303/GM/NIGMS NIH HHS/United States
- R21 OD011196/OD/NIH HHS/United States
- R21 RR032100/RR/NCRR NIH HHS/United States
- R01 GM032443/GM/NIGMS NIH HHS/United States
- R01AR055958/AR/NIAMS NIH HHS/United States
- R01 AR055958/AR/NIAMS NIH HHS/United States
- P01AG033456/AG/NIA NIH HHS/United States
- GM32443S1/GM/NIGMS NIH HHS/United States
- P01 HL098053/HL/NHLBI NIH HHS/United States
- T34 GM008303/GM/NIGMS NIH HHS/United States
- P01HL098053/HL/NHLBI NIH HHS/United States
- R21RR032100/RR/NCRR NIH HHS/United States
- R13 AG033456/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
