

Identification of poly(ADP-ribose) polymerase-1 as a cell cycle regulator through modulating Sp1 mediated transcription in human hepatoma cells
- PMID: 24367566
- PMCID: PMC3868549
- DOI: 10.1371/journal.pone.0082872
Identification of poly(ADP-ribose) polymerase-1 as a cell cycle regulator through modulating Sp1 mediated transcription in human hepatoma cells
Abstract
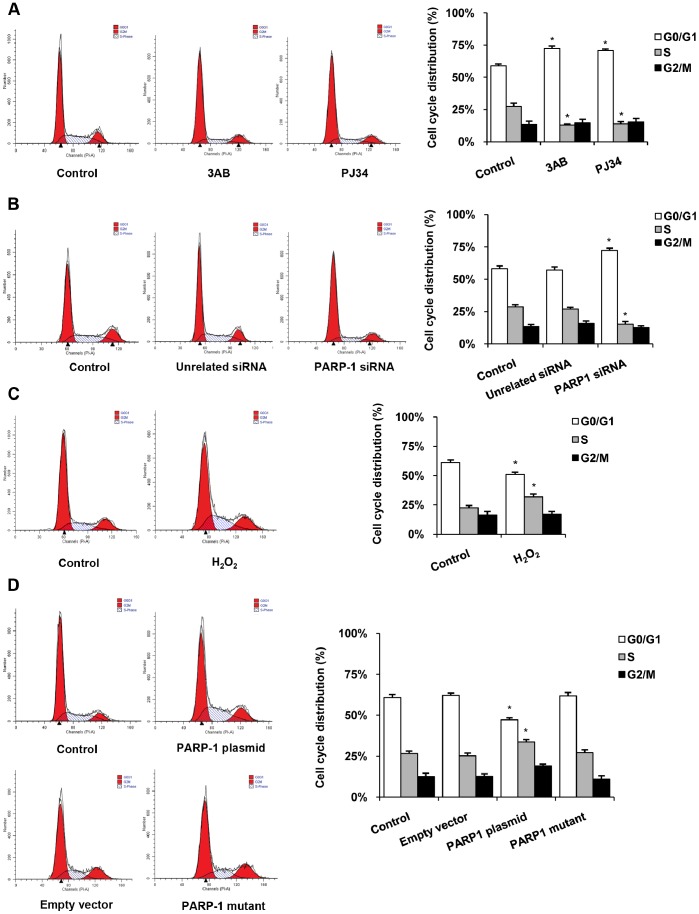
The transcription factor Sp1 is implicated in the activation of G0/G1 phase genes. Modulation of Sp1 transcription activities may affect G1-S checkpoint, resulting in changes in cell proliferation. In this study, our results demonstrated that activated poly(ADP-ribose) polymerase 1 (PARP-1) promoted cell proliferation by inhibiting Sp1 signaling pathway. Cell proliferation and cell cycle assays demonstrated that PARP inhibitors or PARP-1 siRNA treatment significantly inhibited proliferation of hepatoma cells and induced G0/G1 cell cycle arrest in hepatoma cells, while overexpression of PARP-1 or PARP-1 activator treatment promoted cell cycle progression. Simultaneously, inhibition of PARP-1 enhanced the expression of Sp1-mediated checkpoint proteins, such as p21 and p27. In this study, we also showed that Sp1 was poly(ADP-ribosyl)ated by PARP-1 in hepatoma cells. Poly(ADP-ribosyl)ation suppressed Sp1 mediated transcription through preventing Sp1 binding to the Sp1 response element present in the promoters of target genes. Taken together, these data indicated that PARP-1 inhibition attenuated the poly(ADP-ribosyl)ation of Sp1 and significantly increased the expression of Sp1 target genes, resulting in G0/G1 cell cycle arrest and the decreased proliferative ability of the hepatoma cells.
Conflict of interest statement
Figures





References
-
- Kadonaga JT, Carner KR, Masiarz FR, Tjian R (1987) Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell 51: 1079–1090. - PubMed
-
- Black AR, Black JD, Azizkhan-Clifford J (2001) Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol 188: 143–160. - PubMed
-
- Song J, Ugai H, Ogawa K, Wang Y, Sarai A, et al. (2001) Two consecutive zinc fingers in Sp1 and in MAZ are essential for interactions with cis-elements. J Biol Chem 276: 30429–30434. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
