

Neurocognitive and neuropsychiatric phenotypes associated with the mutation L238Q of the α-L-iduronidase gene in Hurler-Scheie syndrome
- PMID: 24368159
- PMCID: PMC3939822
- DOI: 10.1016/j.ymgme.2013.11.014
Neurocognitive and neuropsychiatric phenotypes associated with the mutation L238Q of the α-L-iduronidase gene in Hurler-Scheie syndrome
Abstract
The lysosomal enzyme α-L-iduronidase hydrolyzes terminal iduronic acid from heparan sulfate and dermatan sulfate, and is an essential step in GAG degradation. Mutations of its gene, IDUA, yield a spectrum of mucopolysaccharidosis (MPS) type I clinical disorders. The IDUA mutation, c.712T>A (p.L238Q) was previously noted as a mild mutation. In a longitudinal study of MPS brain structure and function (Lysosomal Disease Network), we found this mutation in 6 of 14 Hurler-Scheie syndrome patients in the age range of 15 to 25 years. We hypothesized that L238Q, when paired with a nonsense mutation, is significantly more severe than other missense-nonsense combinations.
Methods: Of 6 patients with a L238Q mutation, the L238Q allele was paired with a nonsense mutation in 4 patients, paired with a deletion in 1, and with a splice site mutation in another. This group was compared to 6 Hurler-Scheie patients closely matched in age and mutation type. IQ and other neuropsychological tests were administered as part of the protocol. Medical history was compiled into a Physical Symptom Score (PSS). Assessment of IQ, attention, memory, spatial ability, adaptive function and psychological status were measured.
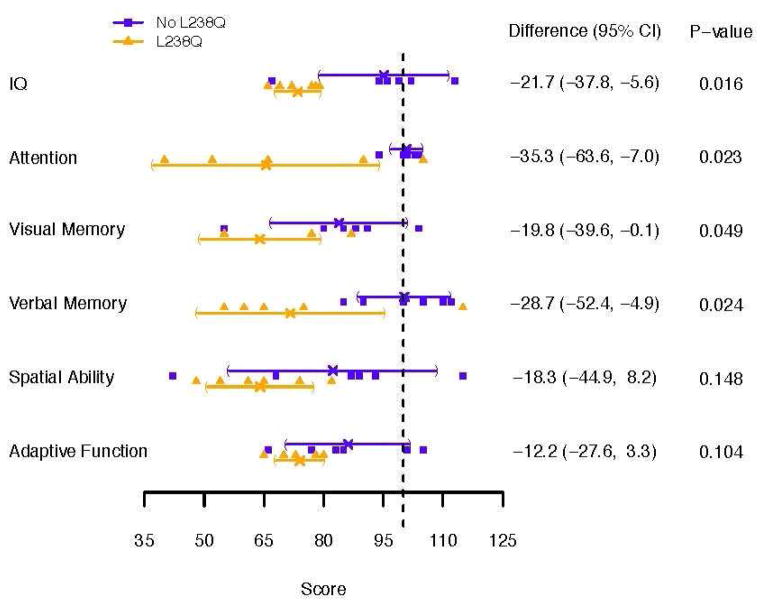
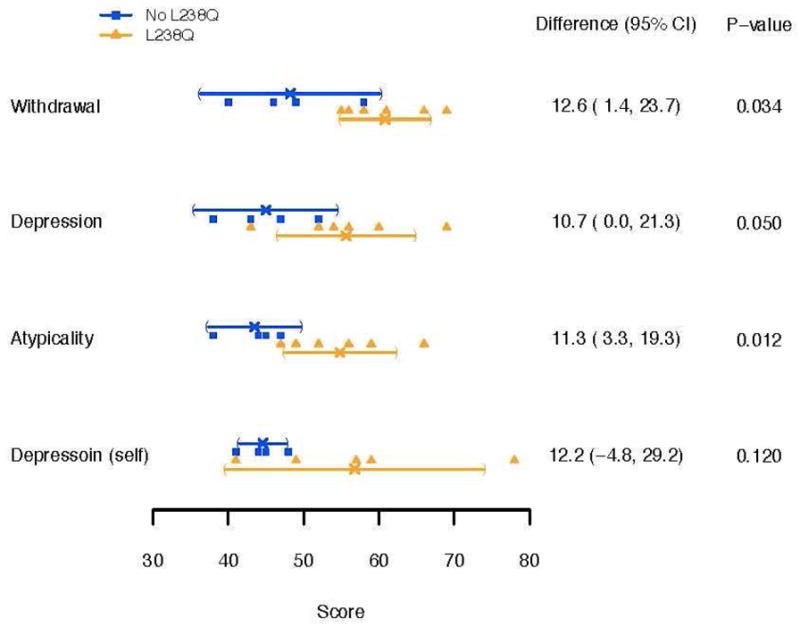
Results: No group differences were found in mean age at evaluation (17.8 and 19.0 years), duration of ERT, or PSS. By history, all were reported to be average in IQ (4/6 with documentation) in early childhood. All (100%) of the L238Q group had a psychiatric history and sleep problems compared to none (0%) of the comparison group. Significant differences were found in depression and withdrawal on parent report measures. IQ was lower in the L238Q group (mean IQ 74) than the comparison group (mean IQ 95; p<0.016). Attention, memory, and visual-spatial ability scores were also significantly lower. Three occurrences of shunted hydrocephalus, and 4 of cervical cord compression were found in the L238Q group; the comparison group had one occurrence of unshunted hydrocephalus and two of cord compression.
Discussion: The missense mutation L238Q, when paired with a nonsense mutation, is associated with significant, late-onset brain disease: psychiatric disorder, cognitive deficit, and general decline starting at a later age than in Hurler syndrome with a mutation-related rate of GAG accumulation and its pathologic sequelae. This particular genotype-phenotype may provide insight into the genesis of psychiatric illnesses more broadly. Consideration of methods for early, brain-targeted treatment in these patients might be considered.
Keywords: Hurler-Scheie syndrome; L238Q; Missense mutation; Mucopolysaccharidosis Type I; Nonsense mutation; α-L-iduronidase.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Pastores GM, Arn P, Beck M, Clarke JTR, Guffon N, Kaplan P, Muenzer J, Norato DYJ, Shapiro E, Thomas J, Viskochil D, Wraith JE. The MPS I registry: Design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Molecular Genetics and Metabolism. 2007;91:37–47. - PubMed
-
- Clarke LA, Heppner J. Mucopolyschharidosis Type I. 2002 Oct 31 (Updated 2011 Jul 21) In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews™ [Internet] Seattle (WA): University of Washington, Seattle; 1993–2013. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1162/
-
- Yogalingam G, Guo XH, Muller VJ, Brooks DA, Clements PR, Kakkis ED, Hopwood JJ. Identification and Molecular Characterization of α-L-Iduronidase Mutations Present in Mucopolysaccharidosis Type I Patients Undergoing Enzyme Replacement Therapy. Human Mutation. 2004;24:199–207. - PubMed
-
- Chandar SS, Mahalingam K. Mucopolysaccharidosis type I: Homology Modeling and docking analysis of the lysosomal enzyme, human α-L-iduronidase. African Journal of Pharmacy and Pharmacology. 2012;6(27):2027–2038.
-
- Saito S, Ohno K, Maita N, Sakuraba H. Structural and clinical implications of amino acid substitutions in α-L-iduronidase: Insight into the basis of mucopolysaccharidosis type I. Molicular Genetics and Metabolism. 2013 in press. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
