

Synaptic dysfunction in prion diseases: a trafficking problem?
- PMID: 24369467
- PMCID: PMC3863542
- DOI: 10.1155/2013/543803
Synaptic dysfunction in prion diseases: a trafficking problem?
Abstract
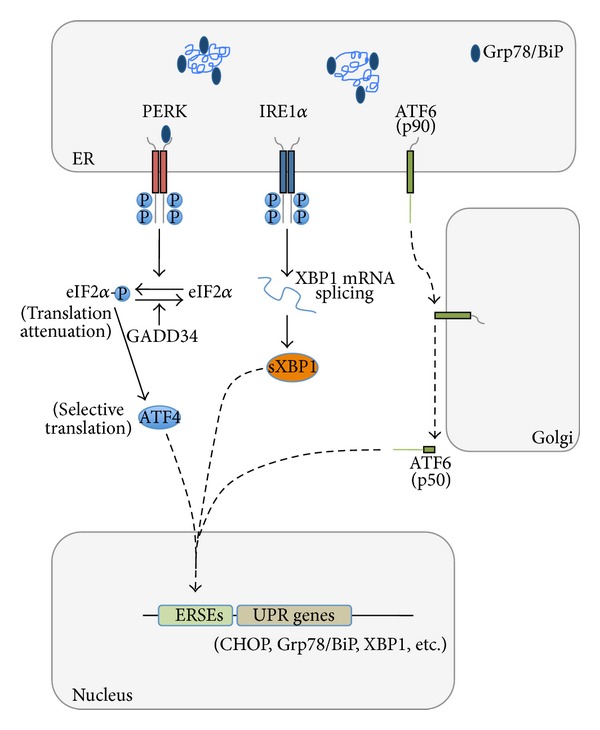
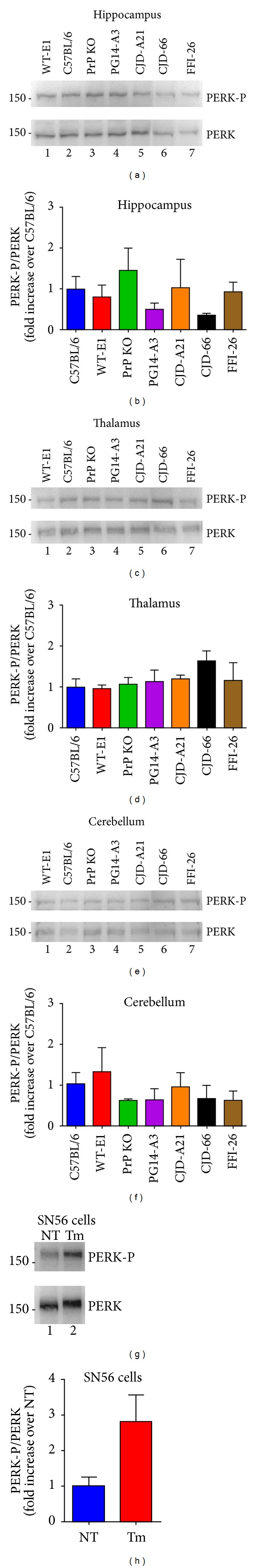
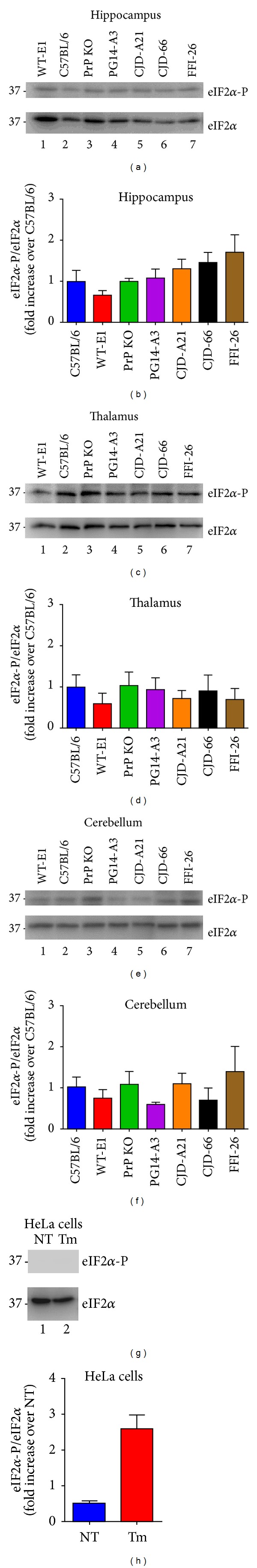
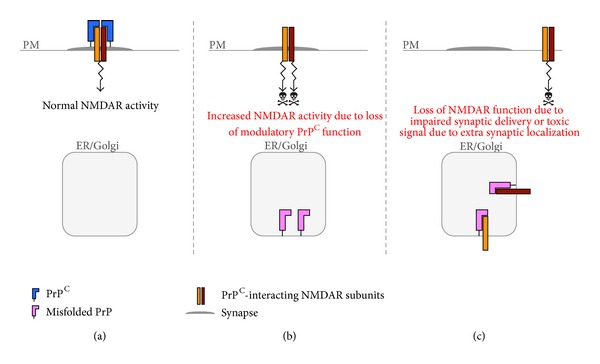
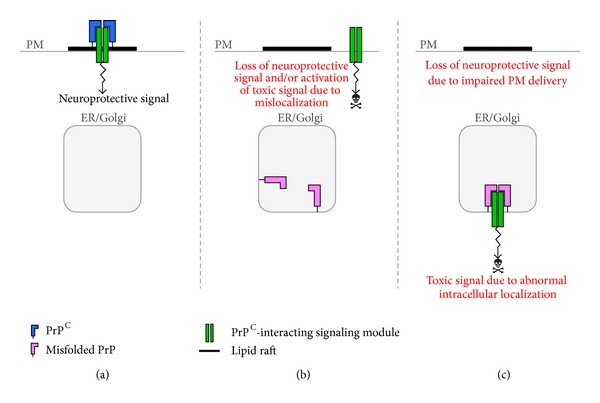
Synaptic dysfunction is an important cause of neurological symptoms in prion diseases, a class of clinically heterogeneous neurodegenerative disorders caused by misfolding of the cellular prion protein (PrP(C)). Experimental data suggest that accumulation of misfolded PrP(C) in the endoplasmic reticulum (ER) may be crucial in synaptic failure, possibly because of the activation of the translational repression pathway of the unfolded protein response. Here, we report that this pathway is not operative in mouse models of genetic prion disease, consistent with our previous observation that ER stress is not involved. Building on our recent finding that ER retention of mutant PrP(C) impairs the secretory trafficking of calcium channels essential for synaptic function, we propose a model of pathogenicity in which intracellular retention of misfolded PrP(C) results in loss of function or gain of toxicity of PrP(C)-interacting proteins. This neurotoxic modality may also explain the phenotypic heterogeneity of prion diseases.
Figures
References
-
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annual Review of Neuroscience. 2001;24:519–550. - PubMed
-
- Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Annals of Neurology. 1994;35(5):513–529. - PubMed
-
- Liberski PP, Sikorska B, Brown P. Kuru: the first prion disease. Advances in Experimental Medicine and Biology. 2012;724:143–153. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
