

Neuroinflammation and copper in Alzheimer's disease
- PMID: 24369524
- PMCID: PMC3863554
- DOI: 10.1155/2013/145345
Neuroinflammation and copper in Alzheimer's disease
Abstract
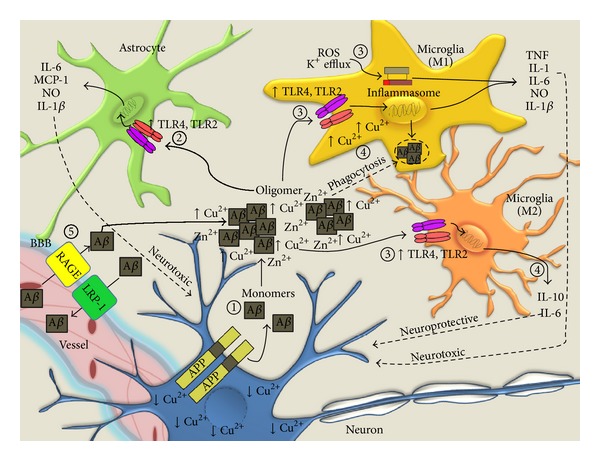
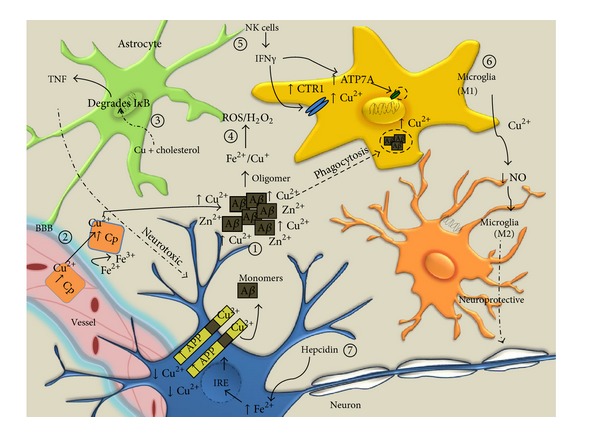
Inflammation is the innate immune response to infection or tissue damage. Initiation of proinflammatory cascades in the central nervous system (CNS) occurs through recognition of danger associated molecular patterns by cognate immune receptors expressed on inflammatory cells and leads to rapid responses to remove the danger stimulus. The presence of activated microglia and astrocytes in the vicinity of amyloid plaques in the brains of Alzheimer's disease (AD) patients and mouse models implicates inflammation as a contributor to AD pathogenesis. Activated microglia play a critical role in amyloid clearance, but chronic deregulation of CNS inflammatory pathways results in secretion of neurotoxic mediators that ultimately contribute to neurodegeneration in AD. Copper (Cu) homeostasis is profoundly affected in AD, and accumulated extracellular Cu drives A β aggregation, while intracellular Cu deficiency limits bioavailable Cu required for CNS functions. This review presents an overview of inflammatory events that occur in AD in response to A β and highlights recent advances on the role of Cu in modulation of beneficial and detrimental inflammatory responses in AD.
Figures
Similar articles
-
Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer's disease.Inflammopharmacology. 2019 Aug;27(4):663-677. doi: 10.1007/s10787-019-00580-x. Epub 2019 Mar 14. Inflammopharmacology. 2019. PMID: 30874945 Review.
-
Type-I interferon pathway in neuroinflammation and neurodegeneration: focus on Alzheimer's disease.J Neural Transm (Vienna). 2018 May;125(5):797-807. doi: 10.1007/s00702-017-1745-4. Epub 2017 Jul 4. J Neural Transm (Vienna). 2018. PMID: 28676934 Review.
-
Neuronal injury in chronic CNS inflammation.Best Pract Res Clin Anaesthesiol. 2010 Dec;24(4):551-62. doi: 10.1016/j.bpa.2010.11.001. Epub 2010 Nov 29. Best Pract Res Clin Anaesthesiol. 2010. PMID: 21619866 Review.
-
Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer's disease.J Neuroinflammation. 2019 Apr 5;16(1):74. doi: 10.1186/s12974-019-1453-0. J Neuroinflammation. 2019. PMID: 30953557 Free PMC article. Review.
-
Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration.Front Cell Neurosci. 2017 Jul 24;11:216. doi: 10.3389/fncel.2017.00216. eCollection 2017. Front Cell Neurosci. 2017. PMID: 28790893 Free PMC article. Review.
Cited by
-
Microglia and Astrocytes in Alzheimer's Disease in the Context of the Aberrant Copper Homeostasis Hypothesis.Biomolecules. 2021 Oct 28;11(11):1598. doi: 10.3390/biom11111598. Biomolecules. 2021. PMID: 34827595 Free PMC article. Review.
-
Morphometric and Nanomechanical Screening of Peripheral Blood Cells with Atomic Force Microscopy for Label-Free Assessment of Alzheimer's Disease, Parkinson's Disease, and Amyotrophic Lateral Sclerosis.Int J Mol Sci. 2023 Sep 19;24(18):14296. doi: 10.3390/ijms241814296. Int J Mol Sci. 2023. PMID: 37762599 Free PMC article. Review.
-
Acute onset neurological symptoms in Wilson disease after traumatic, surgical or emotional events: A cross-sectional study.Medicine (Baltimore). 2019 Jun;98(26):e15917. doi: 10.1097/MD.0000000000015917. Medicine (Baltimore). 2019. PMID: 31261498 Free PMC article.
-
Magnesium Lithospermate B Protects Neurons Against Amyloid β (1-42)-Induced Neurotoxicity Through the NF-κB Pathway.Neurochem Res. 2015 Sep;40(9):1954-65. doi: 10.1007/s11064-015-1691-1. Epub 2015 Aug 19. Neurochem Res. 2015. PMID: 26285901
-
The Effects of Fisetin and Curcumin on Oxidative Damage Caused by Transition Metals in Neurodegenerative Diseases.Mol Neurobiol. 2025 Jan;62(1):1225-1246. doi: 10.1007/s12035-024-04321-2. Epub 2024 Jul 6. Mol Neurobiol. 2025. PMID: 38970766 Review.
References
-
- Fillon M. Details linking chronic inflammation and cancer continue to emerge. Journal of the National Cancer Institute. 2013;105(8):509–510. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
