

Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection
- PMID: 24375466
- PMCID: PMC3999251
- DOI: 10.1002/hep.26976
Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection
Abstract
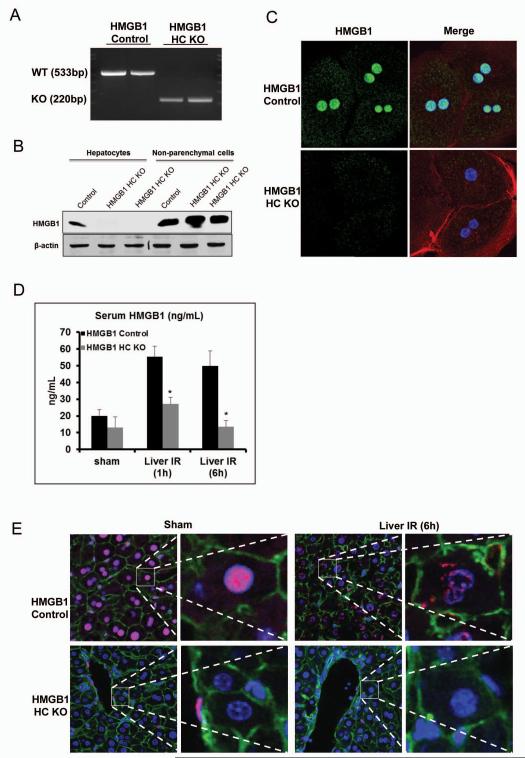
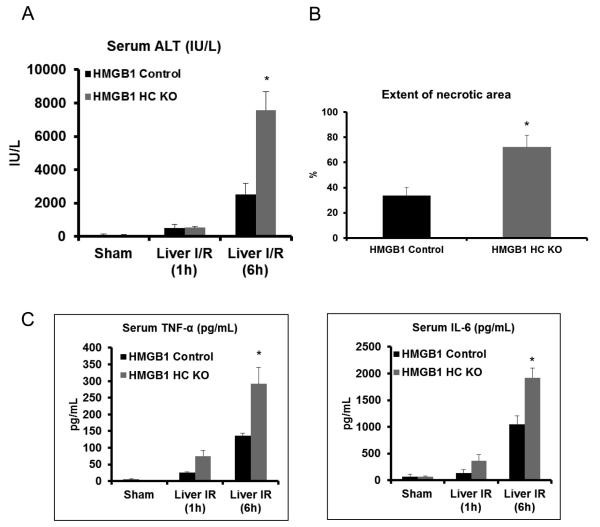
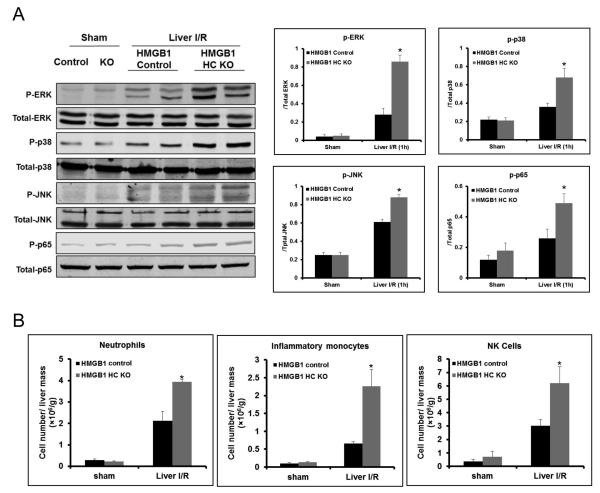
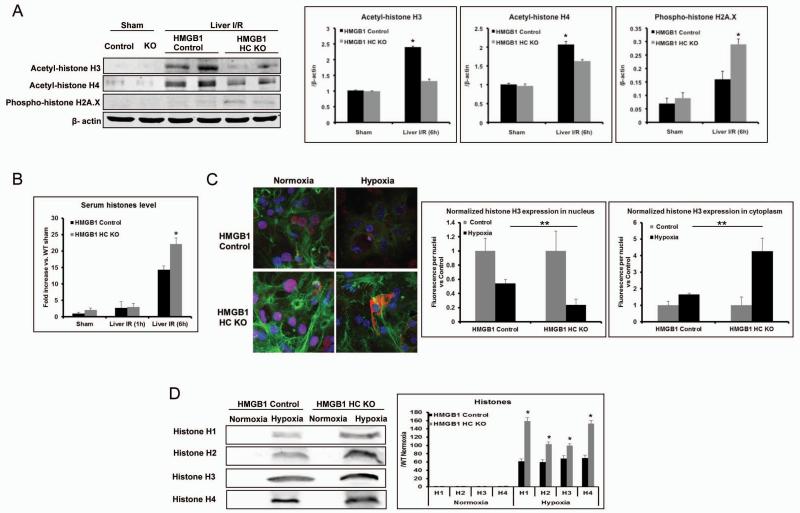
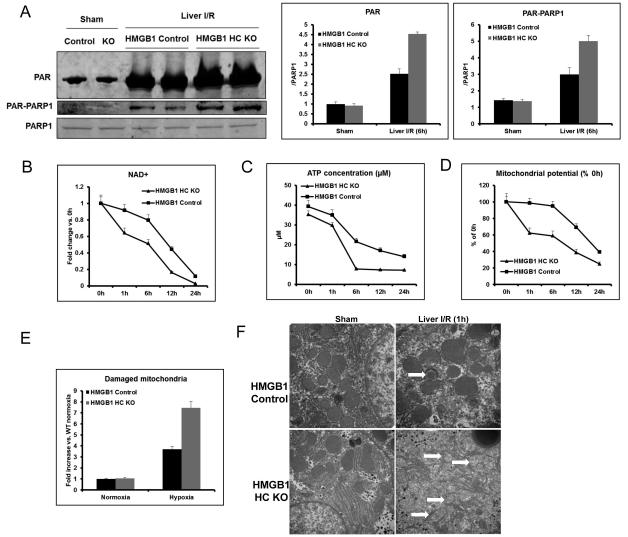
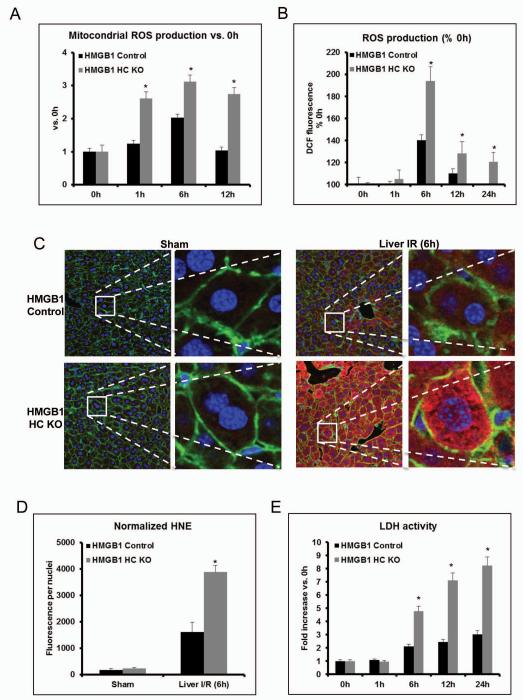
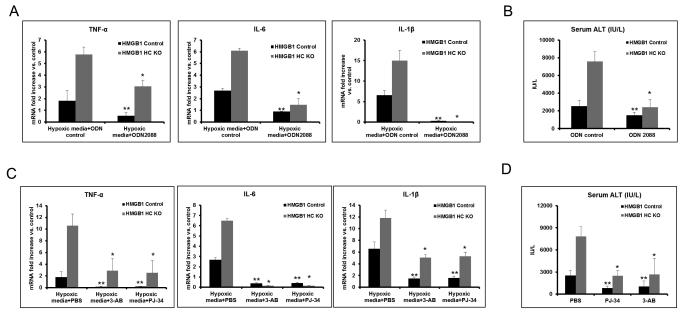
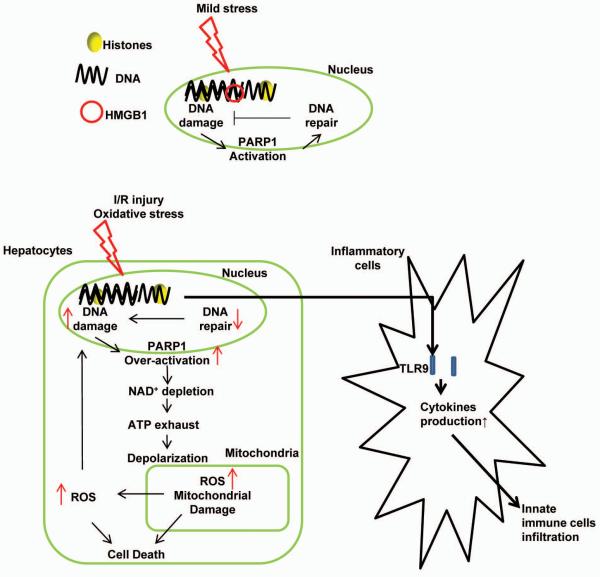
High-mobility group box 1 (HMGB1) is an abundant chromatin-associated nuclear protein and released into the extracellular milieu during liver ischemia-reperfusion (I/R), signaling activation of proinflammatory cascades. Because the intracellular function of HMGB1 during sterile inflammation of I/R is currently unknown, we sought to determine the role of intracellular HMGB1 in hepatocytes after liver I/R. When hepatocyte-specific HMGB1 knockout (HMGB1-HC-KO) and control mice were subjected to a nonlethal warm liver I/R, it was found that HMGB1-HC-KO mice had significantly greater hepatocellular injury after I/R, compared to control mice. Additionally, there was significantly greater DNA damage and decreased chromatin accessibility to repair with lack of HMGB1. Furthermore, lack of hepatocyte HMGB1 led to excessive poly(ADP-ribose)polymerase 1 activation, exhausting nicotinamide adenine dinucleotide and adenosine triphosphate stores, exacerbating mitochondrial instability and damage, and, consequently, leading to increased cell death. We found that this was also associated with significantly more oxidative stress (OS) in HMGB1-HC-KO mice, compared to control. Increased nuclear instability led to a resultant increase in the release of histones with subsequently more inflammatory cytokine production and organ damage through activation of Toll-like receptor 9.
Conclusion: The lack of HMGB1 within hepatocytes leads to increased susceptibility to cellular death after OS conditions.
© 2014 by the American Association for the Study of Liver Diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials