

An extensive evaluation of read trimming effects on Illumina NGS data analysis
- PMID: 24376861
- PMCID: PMC3871669
- DOI: 10.1371/journal.pone.0085024
An extensive evaluation of read trimming effects on Illumina NGS data analysis
Abstract
Next Generation Sequencing is having an extremely strong impact in biological and medical research and diagnostics, with applications ranging from gene expression quantification to genotyping and genome reconstruction. Sequencing data is often provided as raw reads which are processed prior to analysis 1 of the most used preprocessing procedures is read trimming, which aims at removing low quality portions while preserving the longest high quality part of a NGS read. In the current work, we evaluate nine different trimming algorithms in four datasets and three common NGS-based applications (RNA-Seq, SNP calling and genome assembly). Trimming is shown to increase the quality and reliability of the analysis, with concurrent gains in terms of execution time and computational resources needed.
Conflict of interest statement
Figures
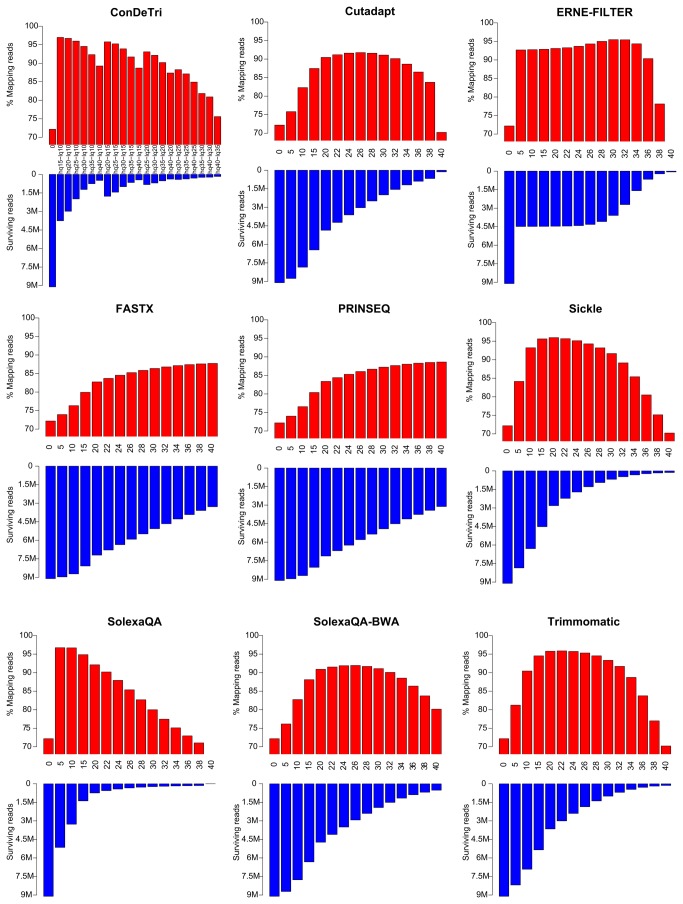

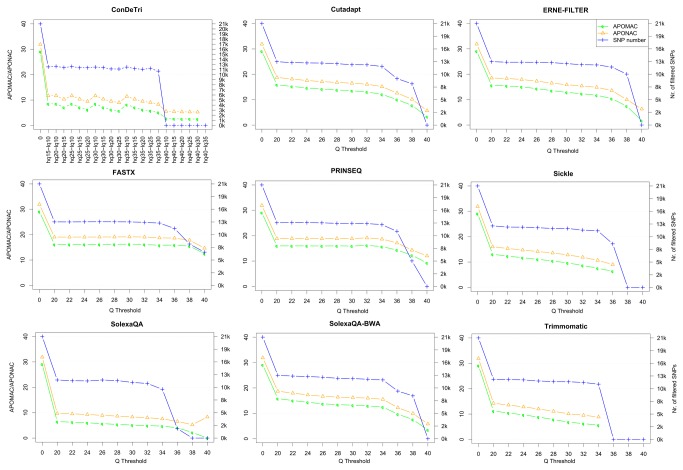

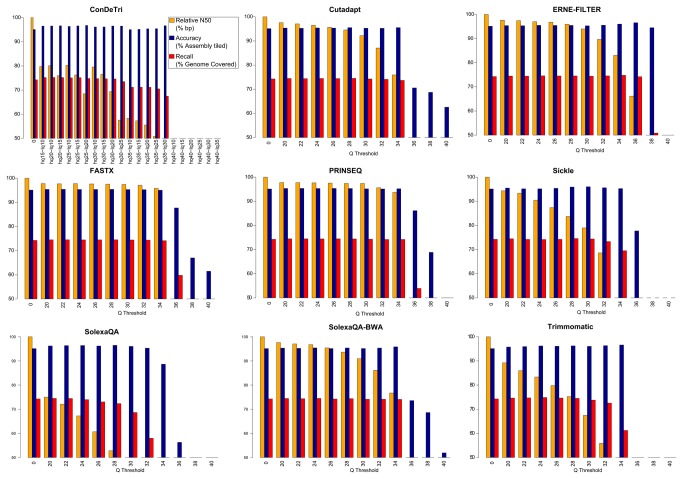
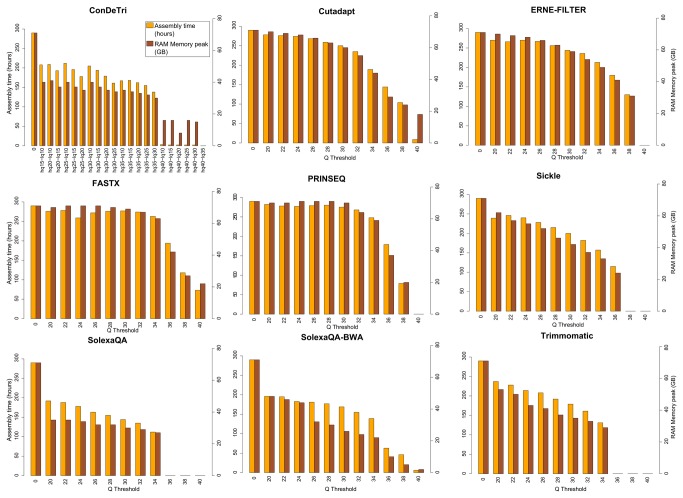
References
-
- Schuster SC (2007) Next-generation sequencing transforms today’s biology. Nature 200. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
