

Synaptic Signaling in Learning and Memory
- PMID: 24379319
- PMCID: PMC4743082
- DOI: 10.1101/cshperspect.a016824
Synaptic Signaling in Learning and Memory
Abstract
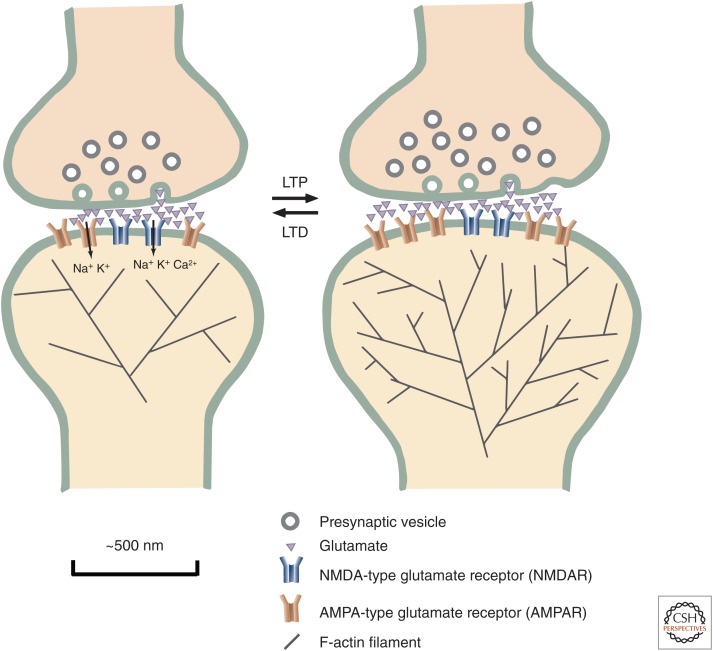
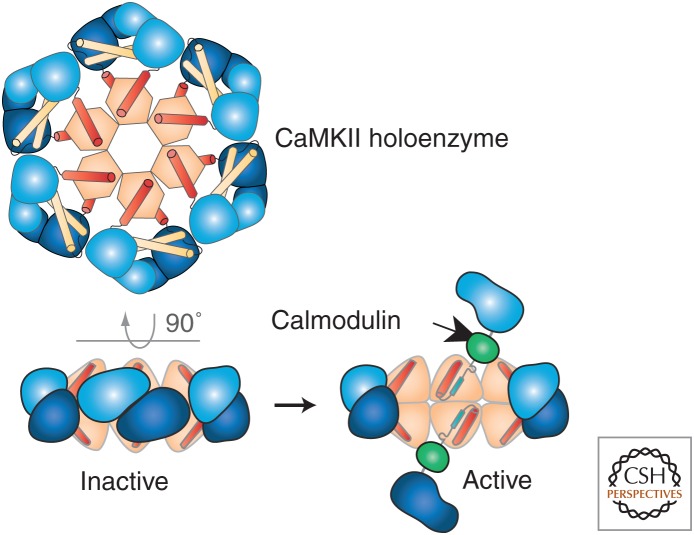
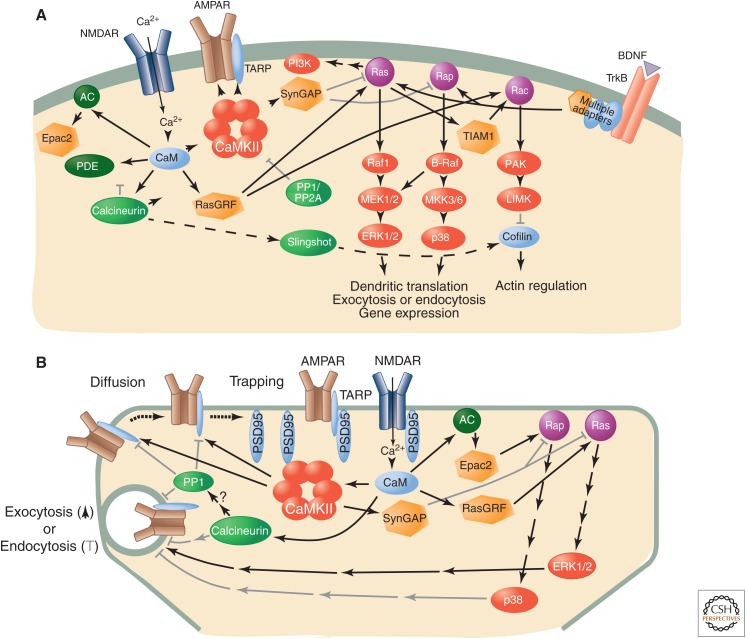
Learning and memory require the formation of new neural networks in the brain. A key mechanism underlying this process is synaptic plasticity at excitatory synapses, which connect neurons into networks. Excitatory synaptic transmission happens when glutamate, the excitatory neurotransmitter, activates receptors on the postsynaptic neuron. Synaptic plasticity is a higher-level process in which the strength of excitatory synapses is altered in response to the pattern of activity at the synapse. It is initiated in the postsynaptic compartment, where the precise pattern of influx of calcium through activated glutamate receptors leads either to the addition of new receptors and enlargement of the synapse (long-term potentiation) or the removal of receptors and shrinkage of the synapse (long-term depression). Calcium/calmodulin-regulated enzymes and small GTPases collaborate to control this highly tuned mechanism.
Copyright © 2016 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures


References
-
- Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. 1993. Modified hippocampal long-term potentiation in PKCγ-mutant mice. Cell 75: 1253–1262. - PubMed
-
- Abraham WC, Bear MF. 1996. Metaplasticity: The plasticity of synaptic plasticity. Trends Neurosci 19: 126–130. - PubMed
-
- Bennett MK, Erondu NE, Kennedy MB. 1983. Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J Biol Chem 258: 12735–12744. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical