

Going through the barrier: coupled disulfide exchange reactions promote efficient catalysis in quiescin sulfhydryl oxidase
- PMID: 24379406
- PMCID: PMC3931083
- DOI: 10.1074/jbc.M113.536219
Going through the barrier: coupled disulfide exchange reactions promote efficient catalysis in quiescin sulfhydryl oxidase
Abstract
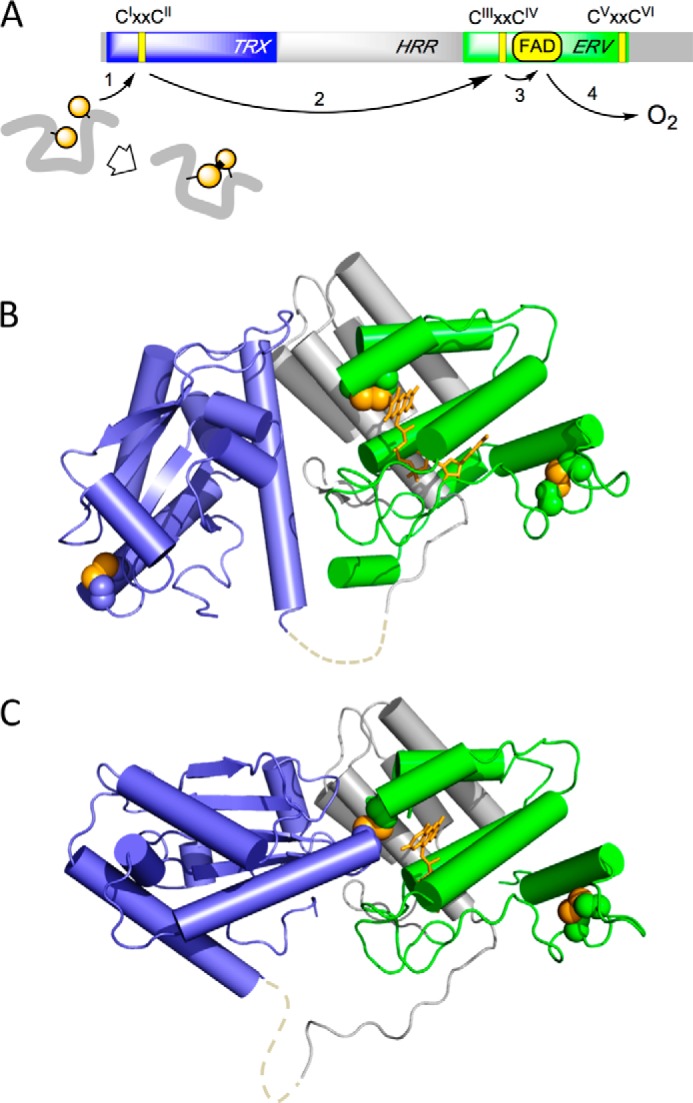
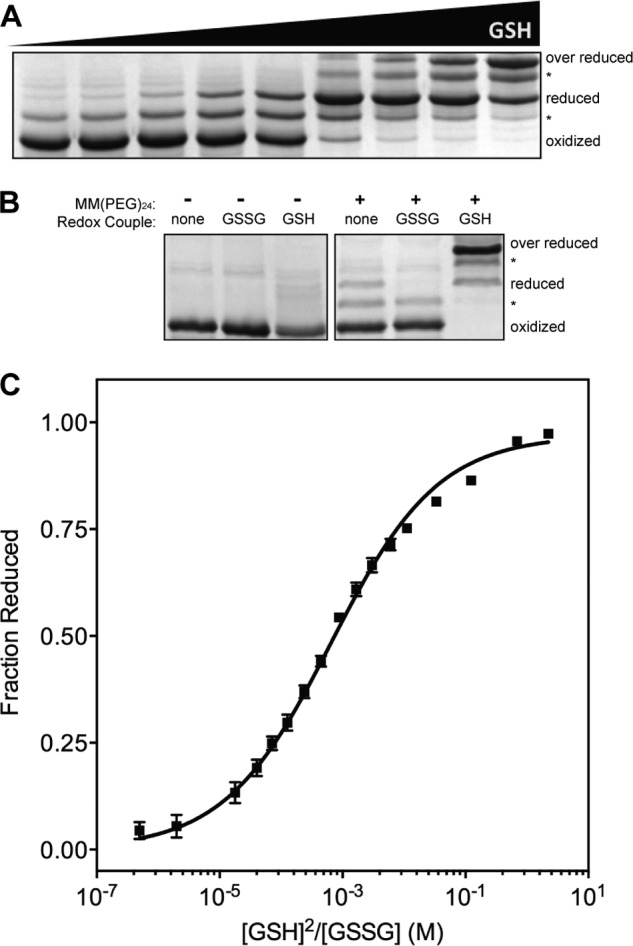
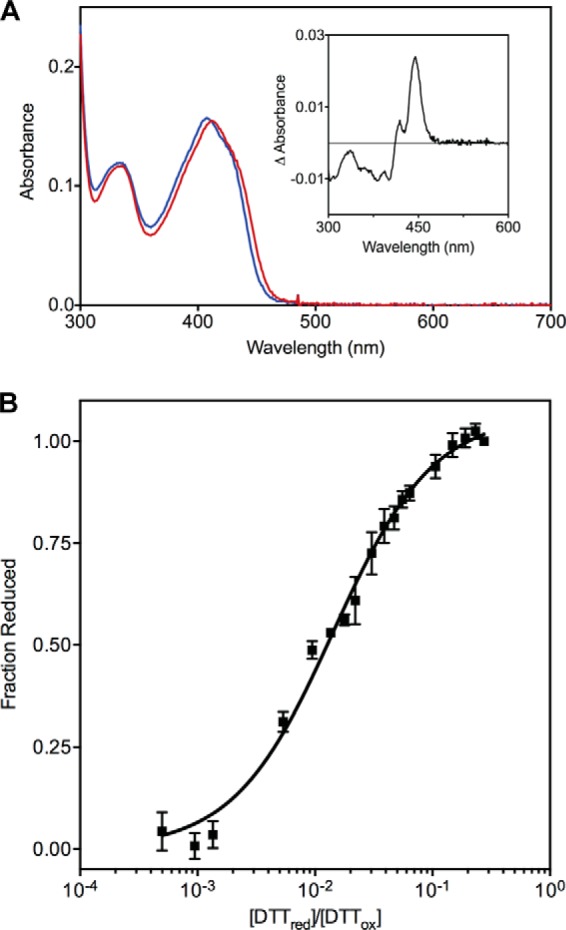
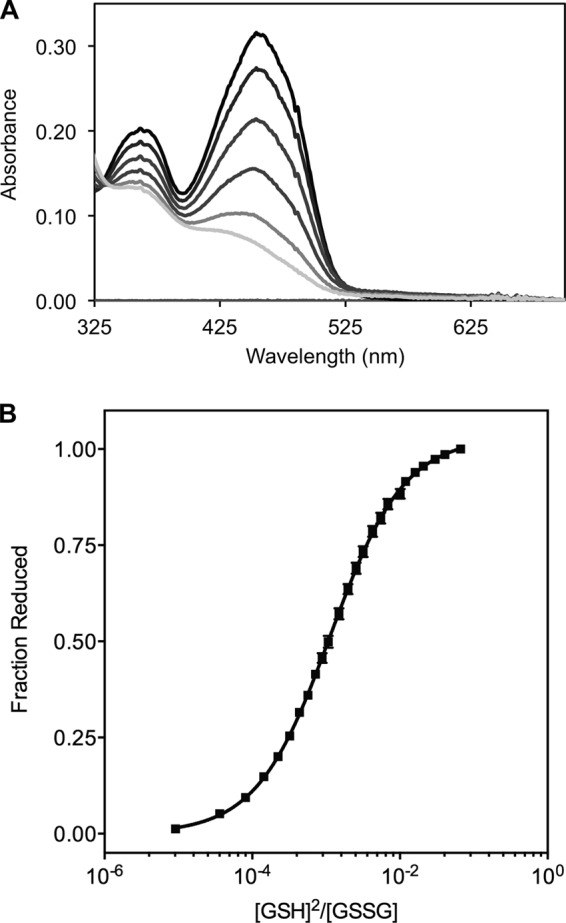
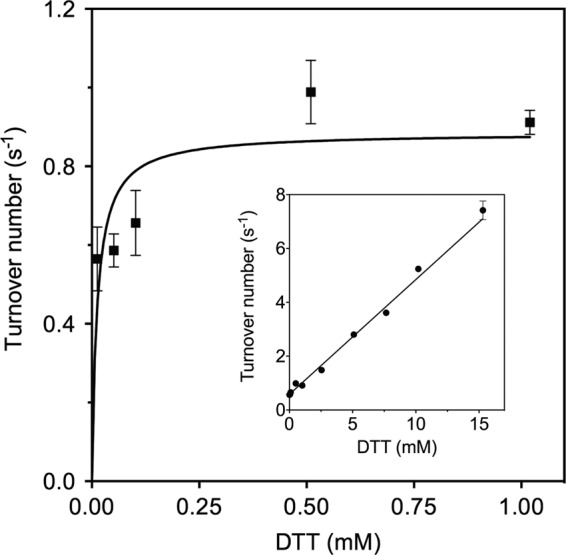
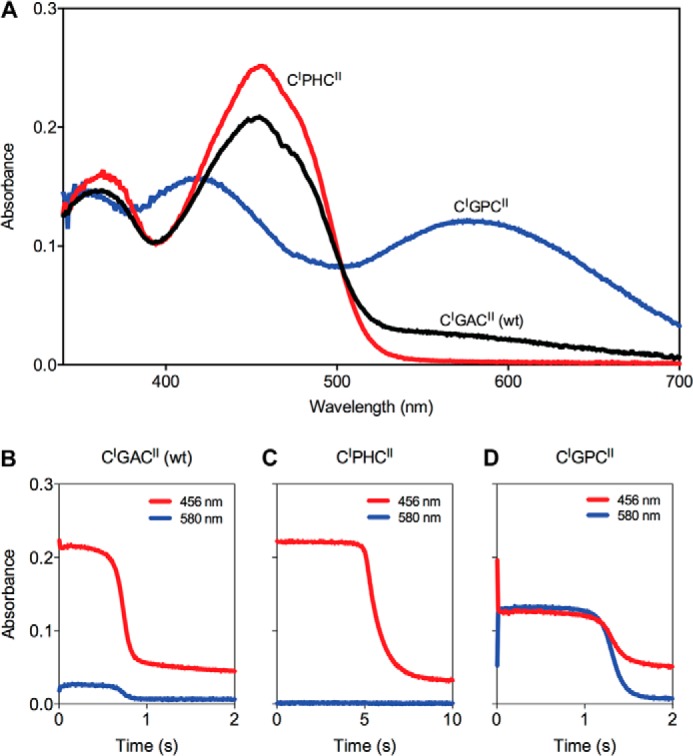
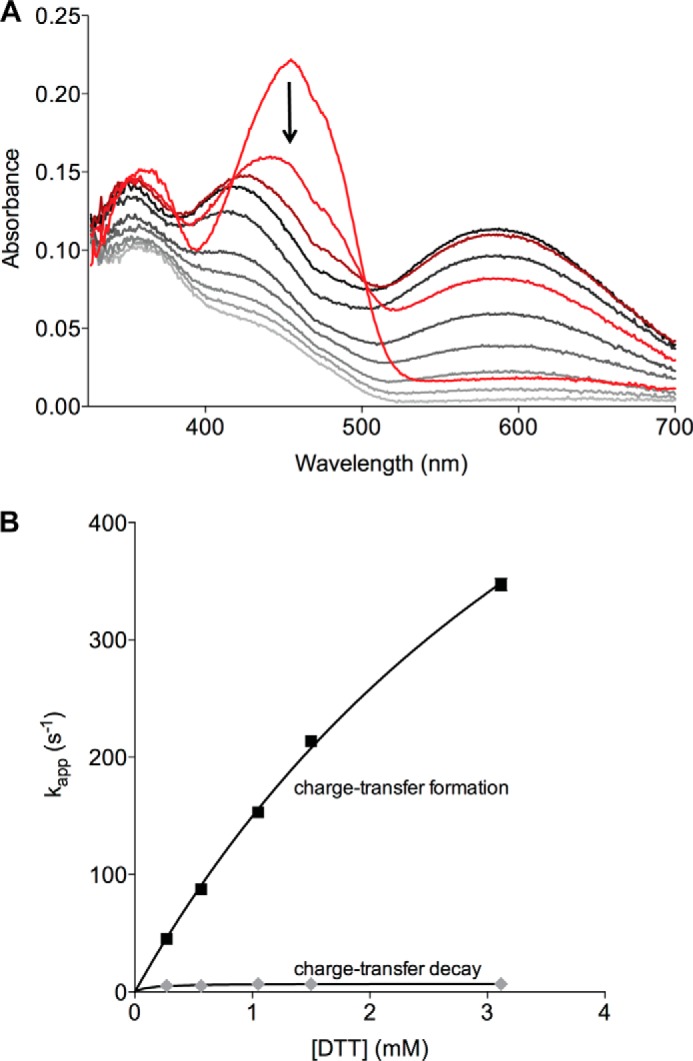
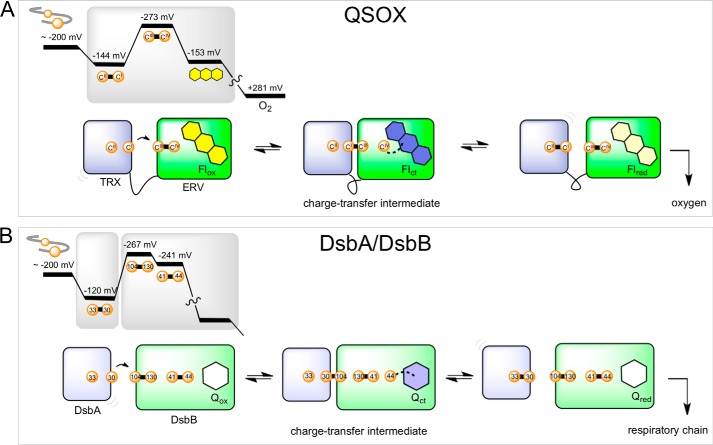
The quiescin sulfhydryl oxidase (QSOX) family of enzymes generates disulfide bonds in peptides and proteins with the reduction of oxygen to hydrogen peroxide. Determination of the potentials of the redox centers in Trypanosoma brucei QSOX provides a context for understanding catalysis by this facile oxidant of protein thiols. The CXXC motif of the thioredoxin domain is comparatively oxidizing (E'0 of -144 mV), consistent with an ability to transfer disulfide bonds to a broad range of thiol substrates. In contrast, the proximal CXXC disulfide in the ERV (essential for respiration and vegetative growth) domain of TbQSOX is strongly reducing (E'0 of -273 mV), representing a major apparent thermodynamic barrier to overall catalysis. Reduction of the oxidizing FAD cofactor (E'0 of -153 mV) is followed by the strongly favorable reduction of molecular oxygen. The role of a mixed disulfide intermediate between thioredoxin and ERV domains was highlighted by rapid reaction studies in which the wild-type CGAC motif in the thioredoxin domain of TbQSOX was replaced by the more oxidizing CPHC or more reducing CGPC sequence. Mixed disulfide bond formation is accompanied by the generation of a charge transfer complex with the flavin cofactor. This provides thermodynamic coupling among the three redox centers of QSOX and avoids the strongly uphill mismatch between the formal potentials of the thioredoxin and ERV disulfides. This work identifies intriguing mechanistic parallels between the eukaryotic QSOX enzymes and the DsbA/B system catalyzing disulfide bond generation in the bacterial periplasm and suggests that the strategy of linked disulfide exchanges may be exploited in other catalysts of oxidative protein folding.
Keywords: Disulfide; Enzyme Mechanisms; Oxidase; Oxidation-Reduction; Protein Folding; QSOX; Quiescin Sulfhydryl Oxidase; Redox; Sulfhydryl.
Figures
Similar articles
-
Quiescin sulfhydryl oxidase from Trypanosoma brucei: catalytic activity and mechanism of a QSOX family member with a single thioredoxin domain.Biochemistry. 2010 Mar 9;49(9):2075-85. doi: 10.1021/bi902222s. Biochemistry. 2010. PMID: 20121244 Free PMC article.
-
Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis.Biochemistry. 2008 Apr 29;47(17):4955-63. doi: 10.1021/bi702522q. Epub 2008 Apr 5. Biochemistry. 2008. PMID: 18393449 Free PMC article.
-
Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: role of a thioredoxin domain in disulfide bond formation.Biochemistry. 2003 Apr 22;42(15):4560-8. doi: 10.1021/bi030003z. Biochemistry. 2003. PMID: 12693953
-
Generating disulfides with the Quiescin-sulfhydryl oxidases.Biochim Biophys Acta. 2008 Apr;1783(4):567-77. doi: 10.1016/j.bbamcr.2007.10.002. Epub 2007 Oct 12. Biochim Biophys Acta. 2008. PMID: 17980160 Free PMC article. Review.
-
Oxidative protein folding and the Quiescin-sulfhydryl oxidase family of flavoproteins.Antioxid Redox Signal. 2010 Oct;13(8):1217-30. doi: 10.1089/ars.2010.3098. Antioxid Redox Signal. 2010. PMID: 20136510 Free PMC article. Review.
Cited by
-
cis-Proline mutants of quiescin sulfhydryl oxidase 1 with altered redox properties undermine extracellular matrix integrity and cell adhesion in fibroblast cultures.Protein Sci. 2019 Jan;28(1):228-238. doi: 10.1002/pro.3537. Protein Sci. 2019. PMID: 30367560 Free PMC article.
-
Redox characterisation of Erv1, a key component for protein import and folding in yeast mitochondria.FEBS J. 2020 Jun;287(11):2281-2291. doi: 10.1111/febs.15136. Epub 2019 Nov 29. FEBS J. 2020. PMID: 31713999 Free PMC article.
-
Oxidative protein folding: from thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum.Free Radic Biol Med. 2015 Mar;80:171-82. doi: 10.1016/j.freeradbiomed.2014.07.037. Epub 2014 Aug 1. Free Radic Biol Med. 2015. PMID: 25091901 Free PMC article. Review.
-
Single-molecule spectroscopy exposes hidden states in an enzymatic electron relay.Nat Commun. 2015 Oct 15;6:8624. doi: 10.1038/ncomms9624. Nat Commun. 2015. PMID: 26468675 Free PMC article.
-
Mia40 is a facile oxidant of unfolded reduced proteins but shows minimal isomerase activity.Arch Biochem Biophys. 2015 Aug 1;579:1-7. doi: 10.1016/j.abb.2015.05.005. Epub 2015 May 23. Arch Biochem Biophys. 2015. PMID: 26014136 Free PMC article.
References
-
- Cabibbo A., Pagani M., Fabbri M., Rocchi M., Farmery M. R., Bulleid N. J., Sitia R. (2000) ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 275, 4827–4833 - PubMed
-
- Pagani M., Fabbri M., Benedetti C., Fassio A., Pilati S., Bulleid N. J., Cabibbo A., Sitia R. (2000) Endoplasmic reticulum oxidoreductin 1-lβ (ERO1-Lβ), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 275, 23685–23692 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
