

Two-phase and family-based designs for next-generation sequencing studies
- PMID: 24379824
- PMCID: PMC3861783
- DOI: 10.3389/fgene.2013.00276
Two-phase and family-based designs for next-generation sequencing studies
Abstract
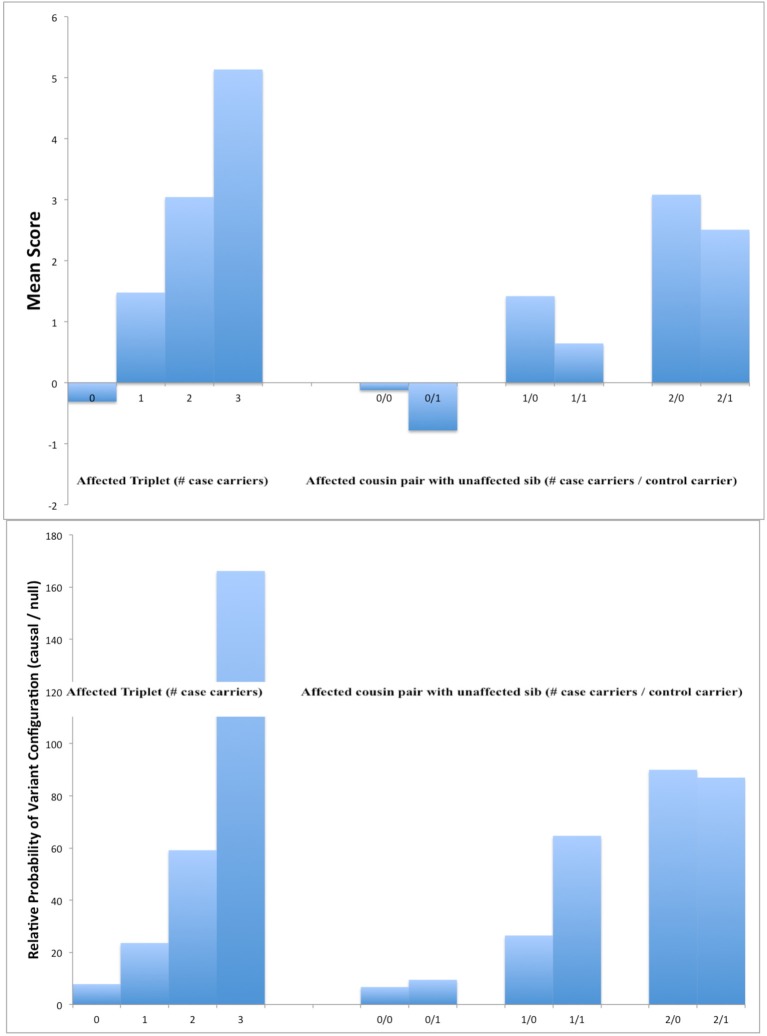
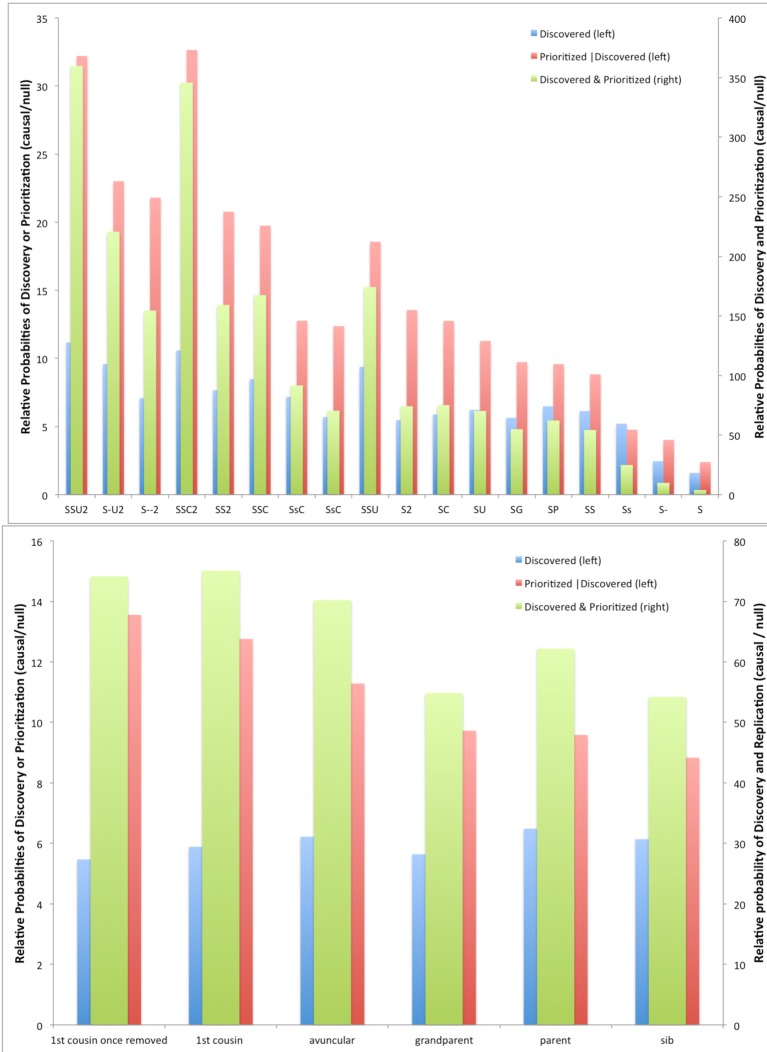
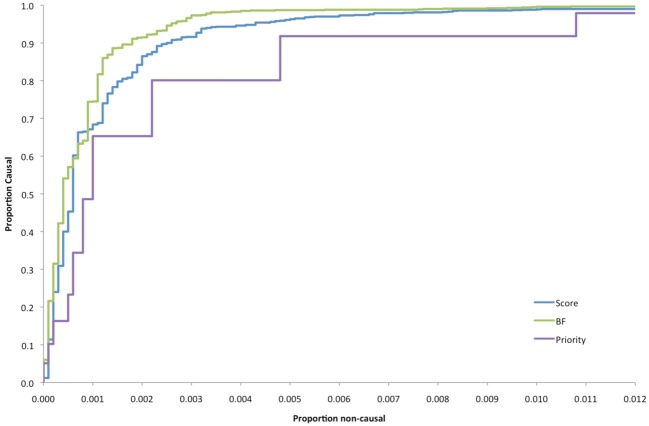
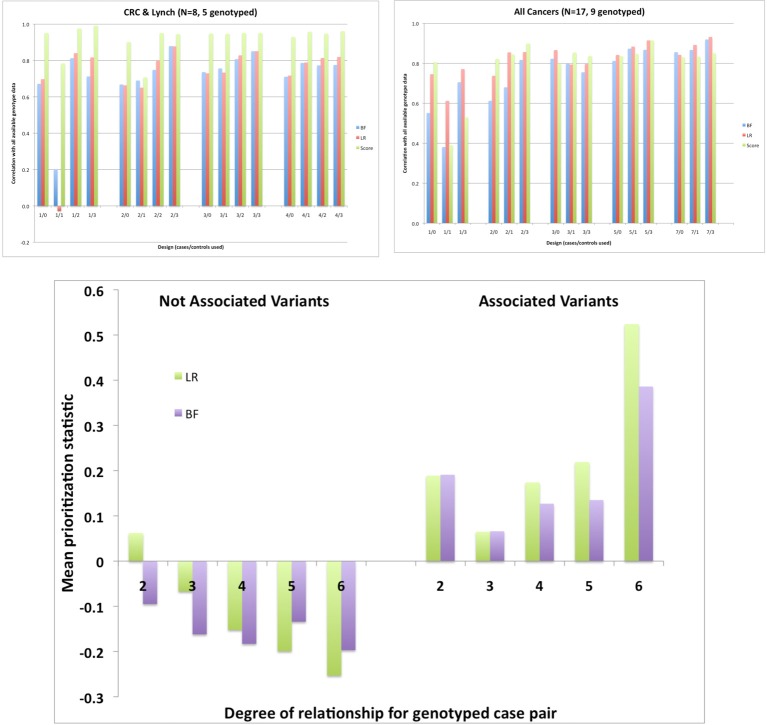
The cost of next-generation sequencing is now approaching that of early GWAS panels, but is still out of reach for large epidemiologic studies and the millions of rare variants expected poses challenges for distinguishing causal from non-causal variants. We review two types of designs for sequencing studies: two-phase designs for targeted follow-up of genomewide association studies using unrelated individuals; and family-based designs exploiting co-segregation for prioritizing variants and genes. Two-phase designs subsample subjects for sequencing from a larger case-control study jointly on the basis of their disease and carrier status; the discovered variants are then tested for association in the parent study. The analysis combines the full sequence data from the substudy with the more limited SNP data from the main study. We discuss various methods for selecting this subset of variants and describe the expected yield of true positive associations in the context of an on-going study of second breast cancers following radiotherapy. While the sharing of variants within families means that family-based designs are less efficient for discovery than sequencing unrelated individuals, the ability to exploit co-segregation of variants with disease within families helps distinguish causal from non-causal ones. Furthermore, by enriching for family history, the yield of causal variants can be improved and use of identity-by-descent information improves imputation of genotypes for other family members. We compare the relative efficiency of these designs with those using unrelated individuals for discovering and prioritizing variants or genes for testing association in larger studies. While associations can be tested with single variants, power is low for rare ones. Recent generalizations of burden or kernel tests for gene-level associations to family-based data are appealing. These approaches are illustrated in the context of a family-based study of colorectal cancer.
Keywords: breast neoplasms; colorectal cancer; family-based study; rare variant association; sequencing; two-phase sampling design.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases