

Computational methods in drug discovery
- PMID: 24381236
- PMCID: PMC3880464
- DOI: 10.1124/pr.112.007336
Computational methods in drug discovery
Abstract
Computer-aided drug discovery/design methods have played a major role in the development of therapeutically important small molecules for over three decades. These methods are broadly classified as either structure-based or ligand-based methods. Structure-based methods are in principle analogous to high-throughput screening in that both target and ligand structure information is imperative. Structure-based approaches include ligand docking, pharmacophore, and ligand design methods. The article discusses theory behind the most important methods and recent successful applications. Ligand-based methods use only ligand information for predicting activity depending on its similarity/dissimilarity to previously known active ligands. We review widely used ligand-based methods such as ligand-based pharmacophores, molecular descriptors, and quantitative structure-activity relationships. In addition, important tools such as target/ligand data bases, homology modeling, ligand fingerprint methods, etc., necessary for successful implementation of various computer-aided drug discovery/design methods in a drug discovery campaign are discussed. Finally, computational methods for toxicity prediction and optimization for favorable physiologic properties are discussed with successful examples from literature.
Figures
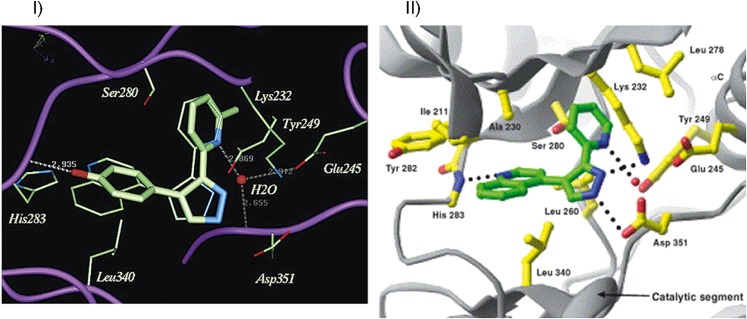
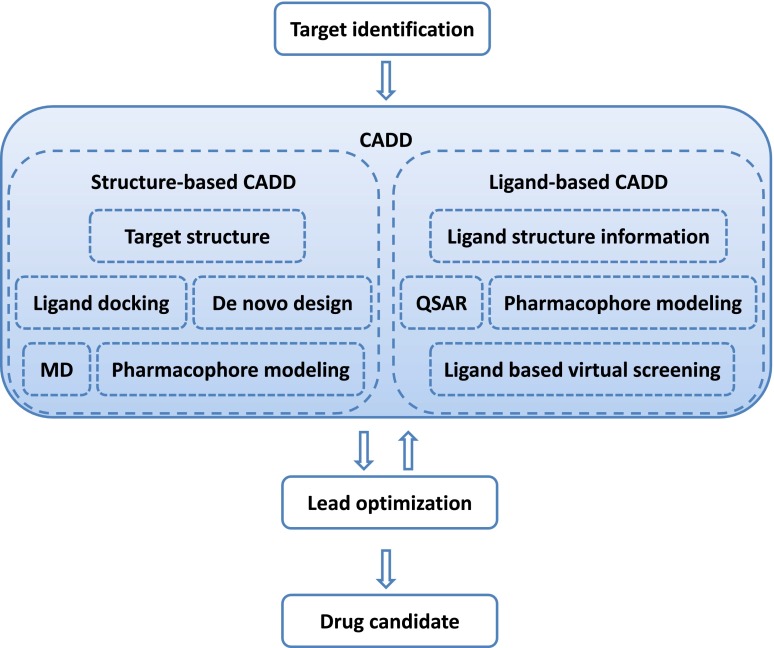
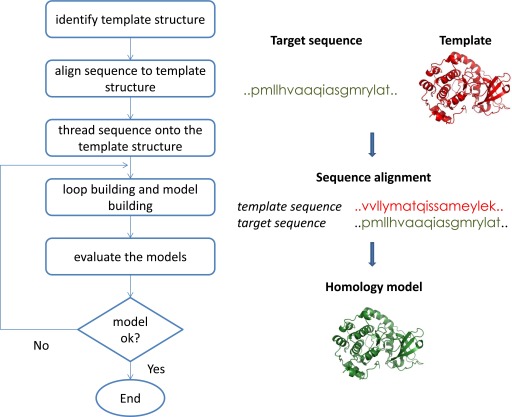
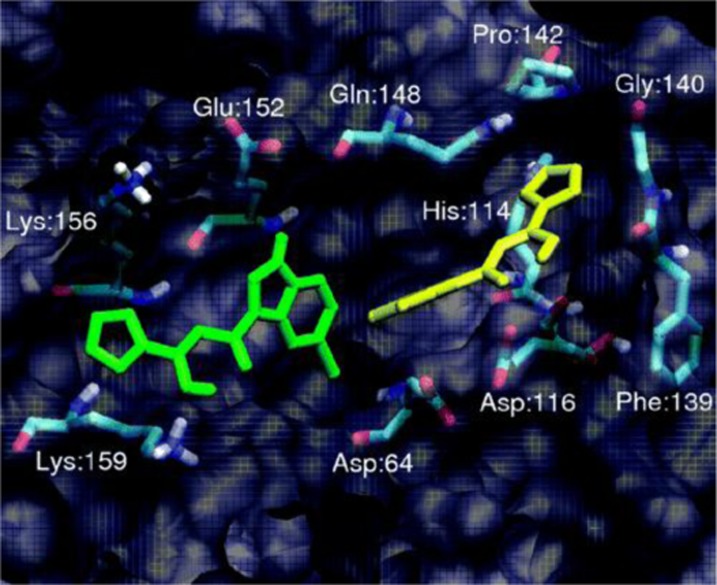
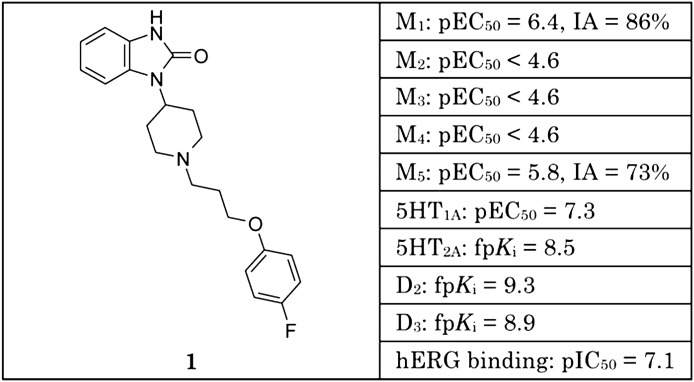
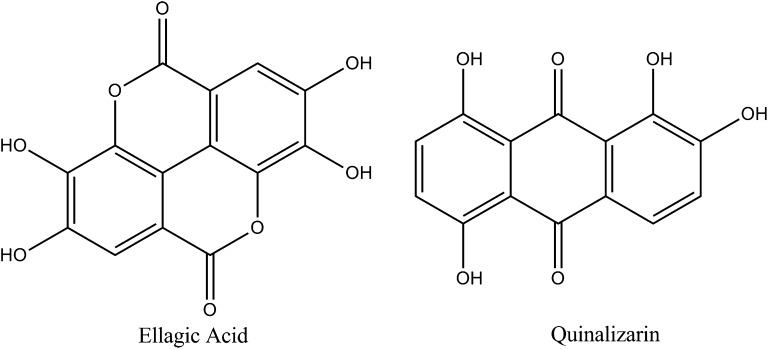
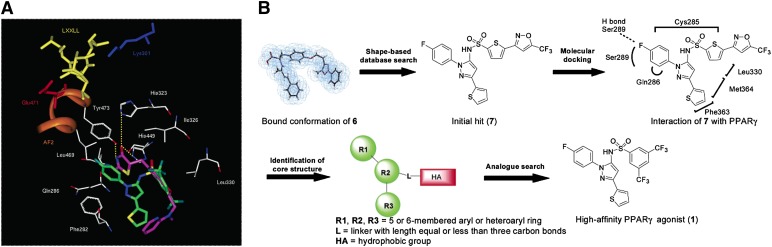
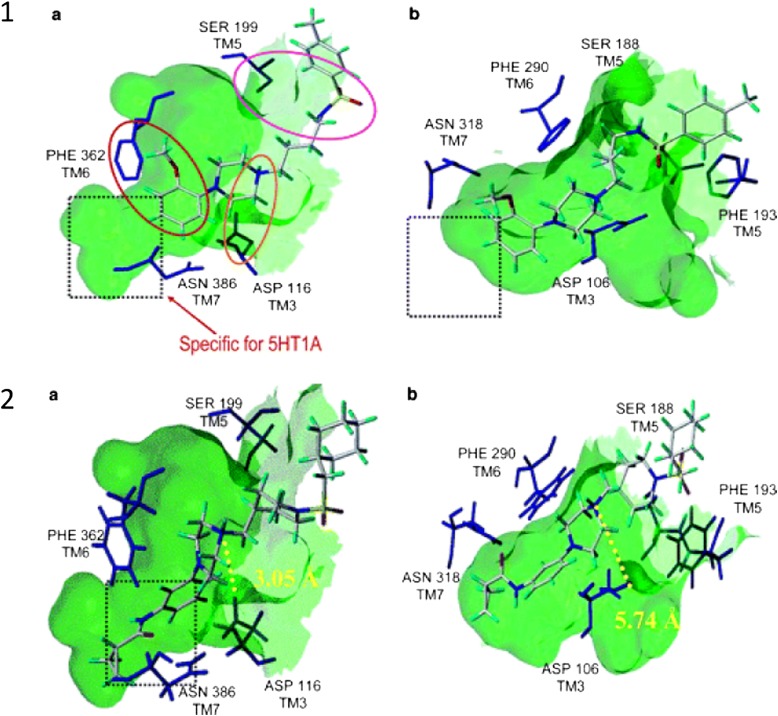
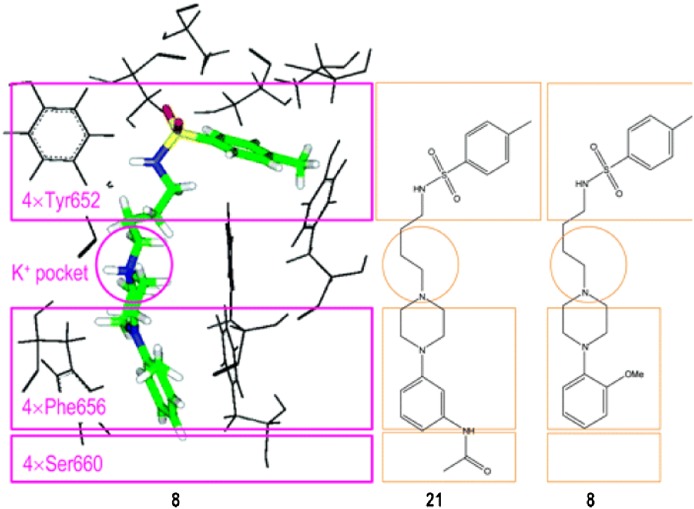
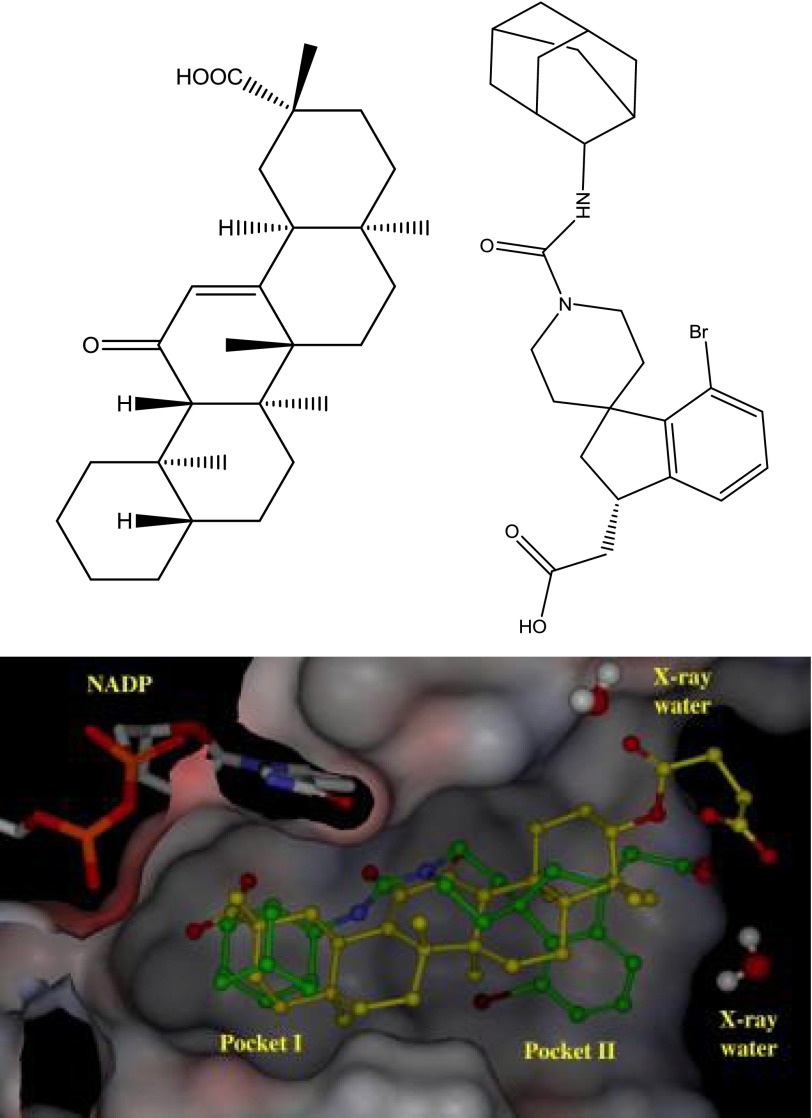
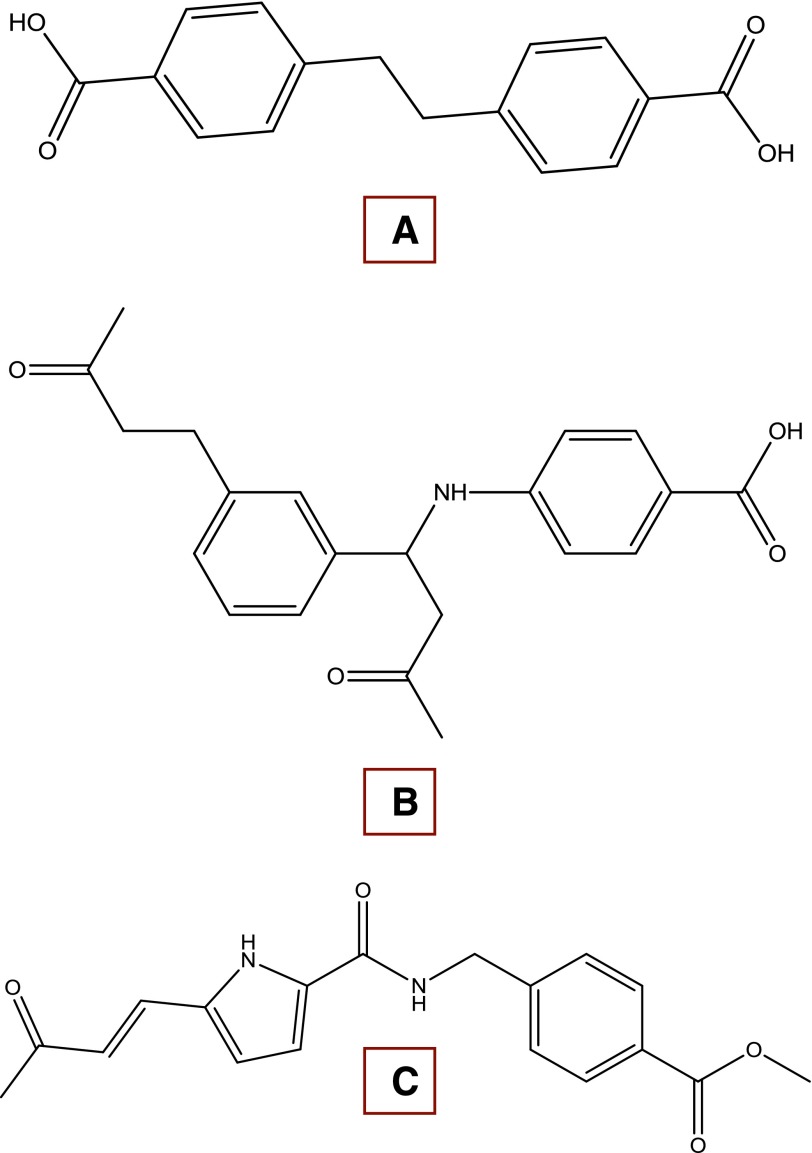
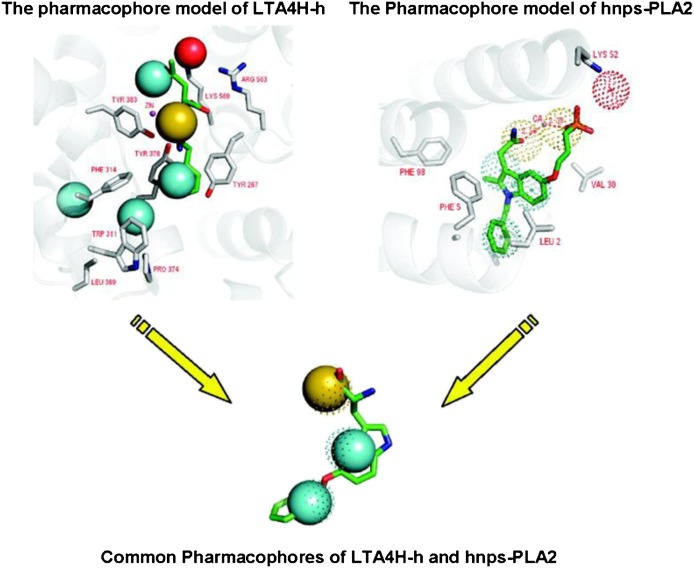
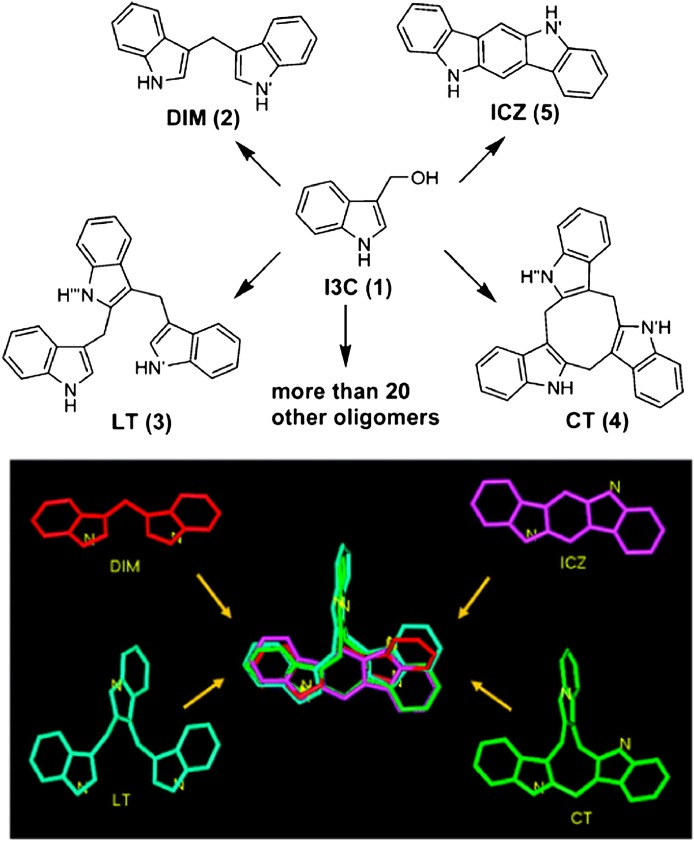
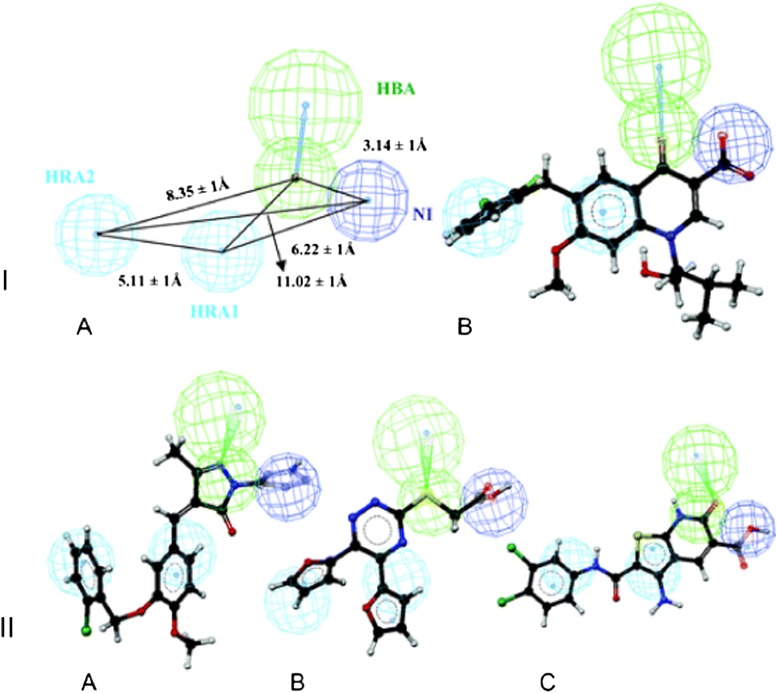
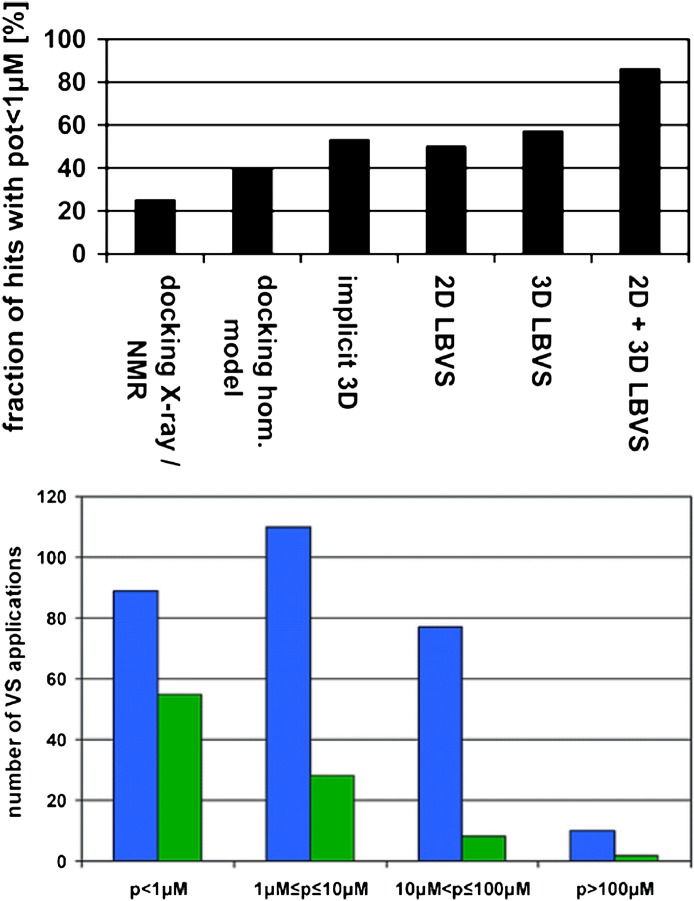
References
-
- Abagyan R, Lee WH, Raush E, Budagyan L, Totrov M, Sundstrom M, Marsden BD. (2006) Disseminating structural genomics data to the public: from a data dump to an animated story. Trends Biochem Sci 31:76–78 - PubMed
-
- Abagyan R, Totrov M,, Kuznetsov D. (1994) ICM - a new method for protein modeling and design–applications to docking and structure prediction from the distorted native conformation. J Comput Chem 15:488–506
-
- Accelrys (2013) Accelrys metabolite. Available from http://accelrys.com/products/databases/bioactivity/metabolite.html
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
