

Progressive myoclonic epilepsies: definitive and still undetermined causes
- PMID: 24384641
- PMCID: PMC3917687
- DOI: 10.1212/WNL.0000000000000077
Progressive myoclonic epilepsies: definitive and still undetermined causes
Abstract
Objective: To define the clinical spectrum and etiology of progressive myoclonic epilepsies (PMEs) in Italy using a database developed by the Genetics Commission of the Italian League against Epilepsy.
Methods: We collected clinical and laboratory data from patients referred to 25 Italian epilepsy centers regardless of whether a positive causative factor was identified. PMEs of undetermined origins were grouped using 2-step cluster analysis.
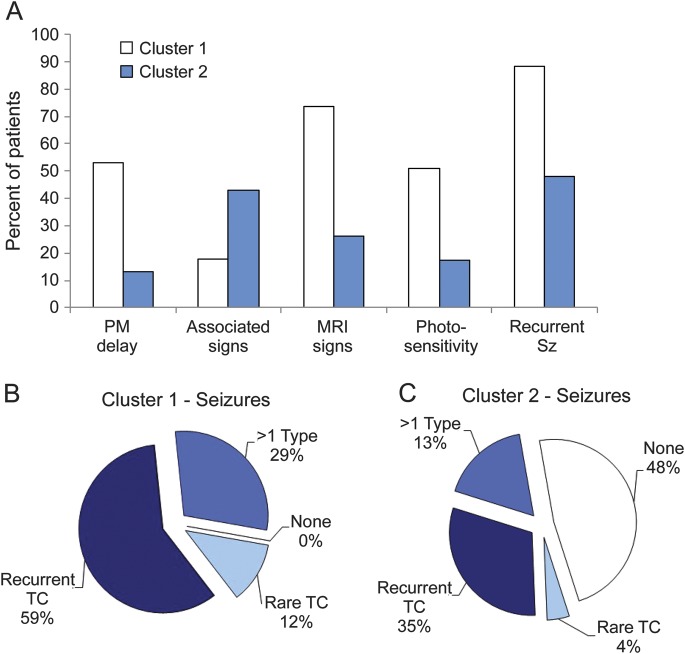
Results: We collected clinical data from 204 patients, including 77 with a diagnosis of Unverricht-Lundborg disease and 37 with a diagnosis of Lafora body disease; 31 patients had PMEs due to rarer genetic causes, mainly neuronal ceroid lipofuscinoses. Two more patients had celiac disease. Despite extensive investigation, we found no definitive etiology for 57 patients. Cluster analysis indicated that these patients could be grouped into 2 clusters defined by age at disease onset, age at myoclonus onset, previous psychomotor delay, seizure characteristics, photosensitivity, associated signs other than those included in the cardinal definition of PME, and pathologic MRI findings.
Conclusions: Information concerning the distribution of different genetic causes of PMEs may provide a framework for an updated diagnostic workup. Phenotypes of the patients with PME of undetermined cause varied widely. The presence of separate clusters suggests that novel forms of PME are yet to be clinically and genetically characterized.
Figures


Comment in
-
Progressive myoclonic epilepsies: it takes a village to make a diagnosis.Neurology. 2014 Feb 4;82(5):378-9. doi: 10.1212/WNL.0000000000000091. Epub 2014 Jan 2. Neurology. 2014. PMID: 24384640
References
-
- de Siqueira LF. Progressive myoclonic epilepsies: review of clinical, molecular and therapeutic aspects. J Neurol 2010;257:1612–1619 - PubMed
-
- Dibbens LM, Michelucci R, Gambardella A, et al. SCARB2 mutations in progressive myoclonus epilepsy (PME) without renal failure. Ann Neurol 2009;66:532–536 - PubMed
-
- Berkovic SF, Andermann F, Carpenter S, Wolfe LS. Progressive myoclonus epilepsies: specific causes and diagnosis. N Engl J Med 1986;315:296–305 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials