

Dysbindin is a potent inducer of RhoA-SRF-mediated cardiomyocyte hypertrophy
- PMID: 24385487
- PMCID: PMC3840930
- DOI: 10.1083/jcb.201303052
Dysbindin is a potent inducer of RhoA-SRF-mediated cardiomyocyte hypertrophy
Abstract
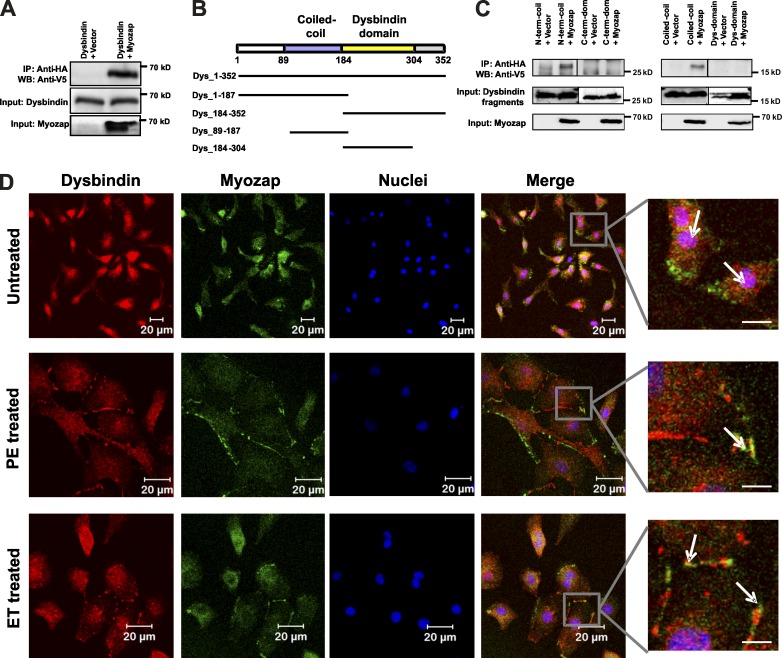
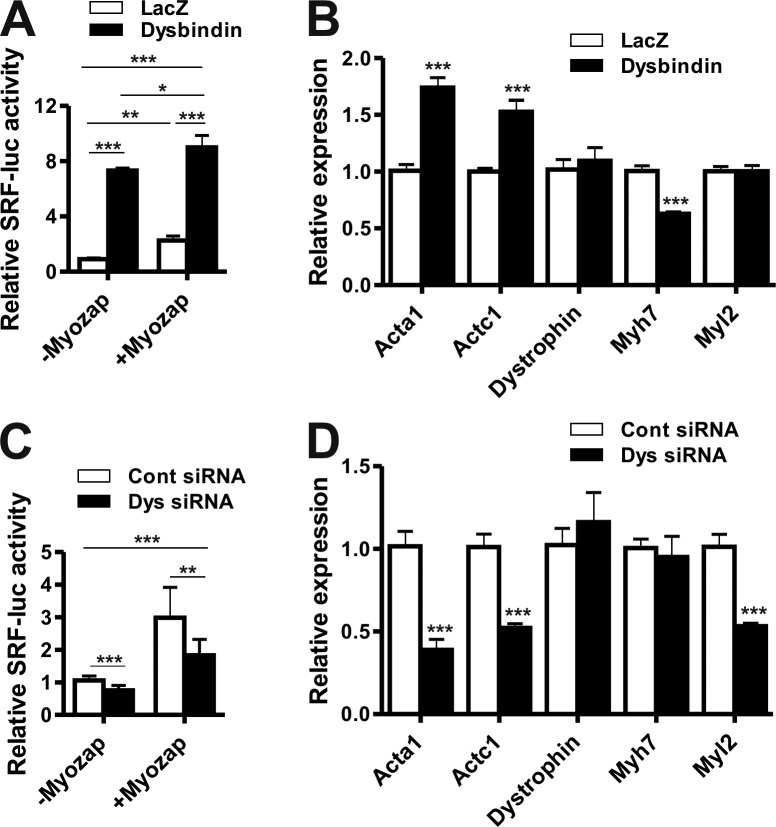
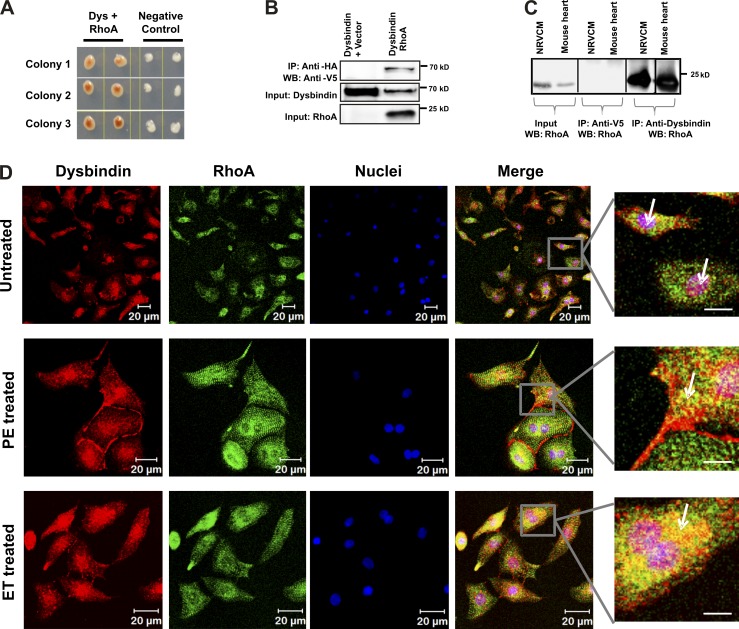
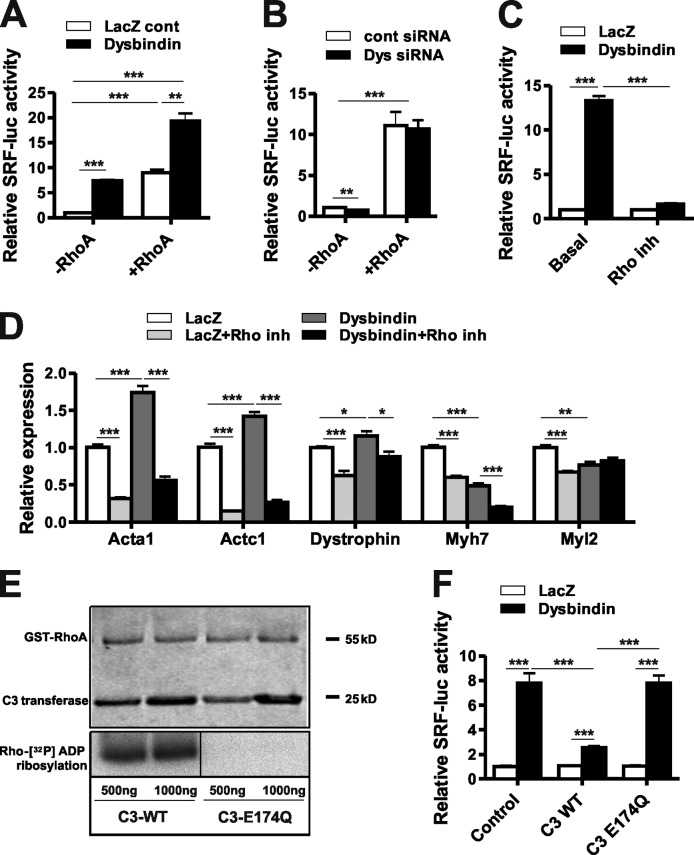
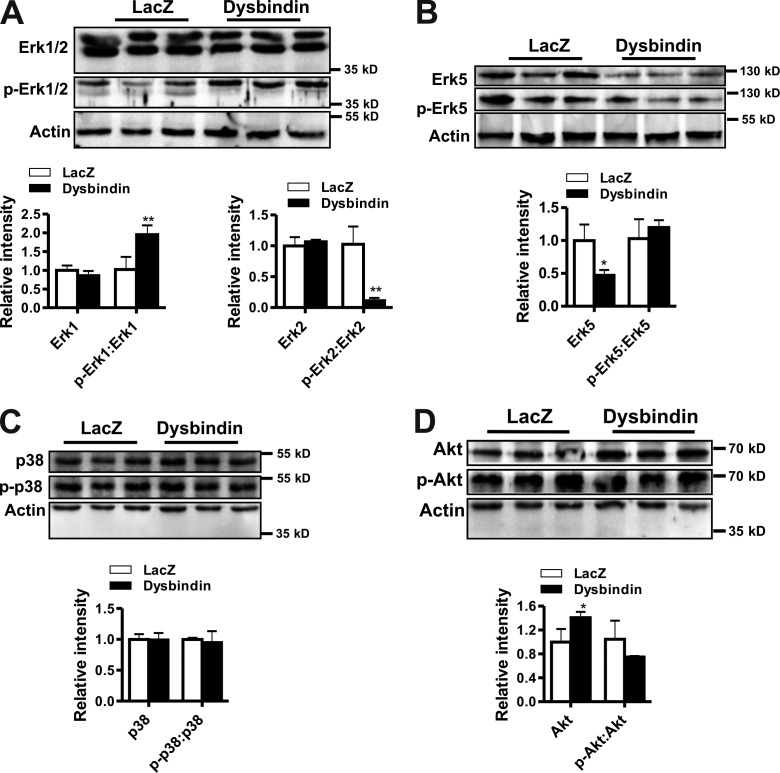
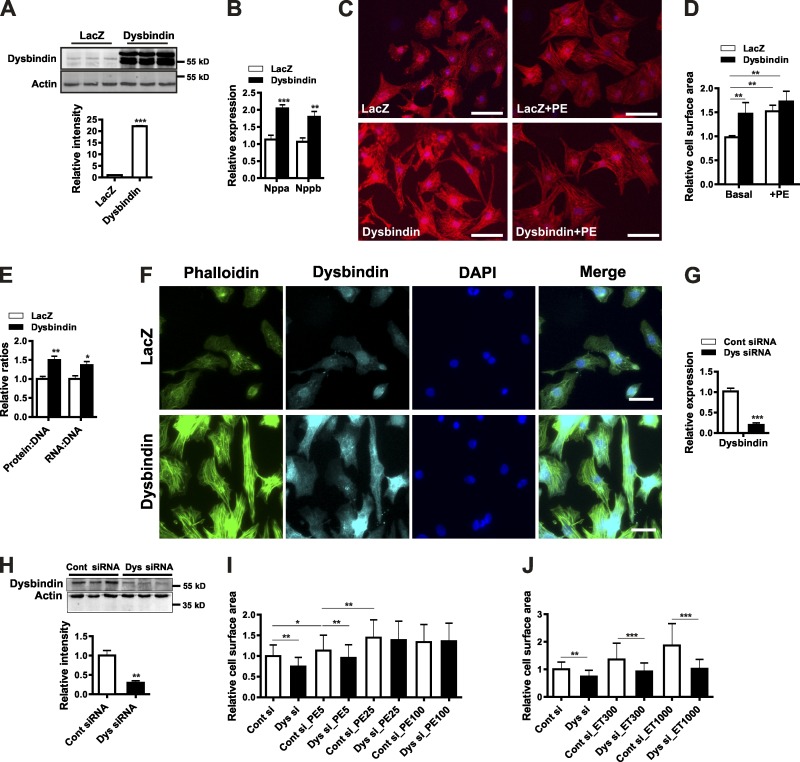
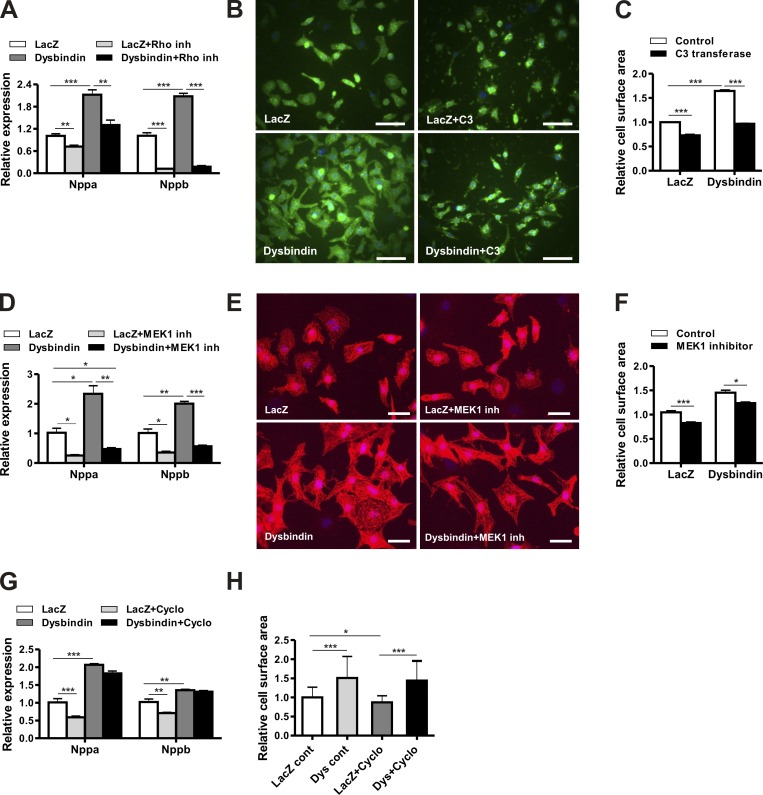
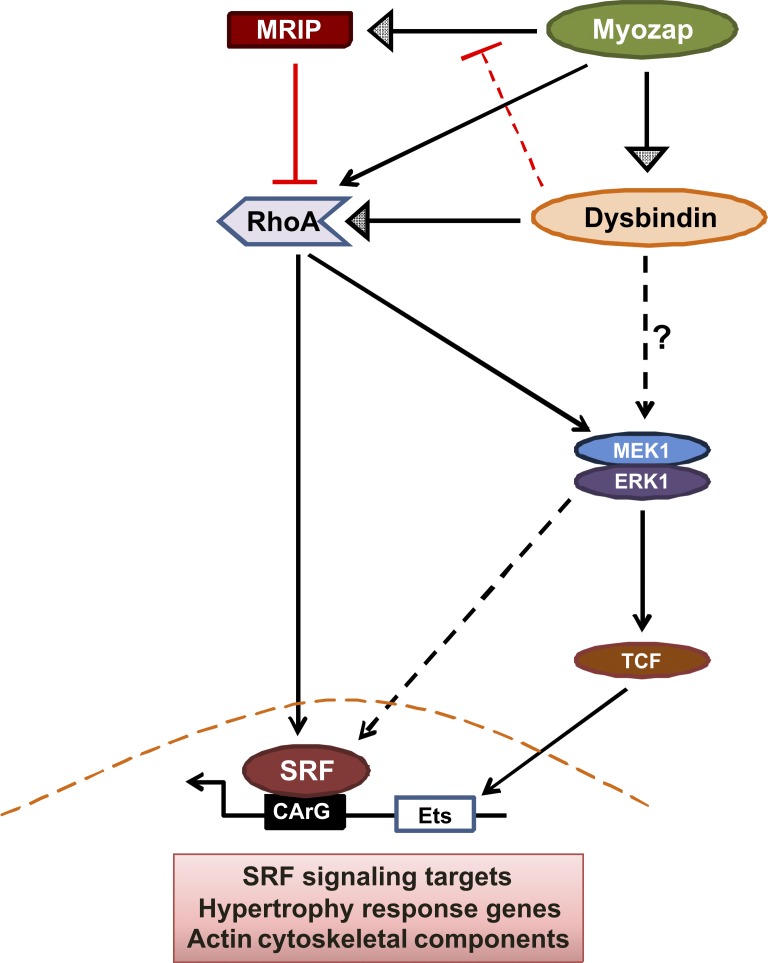
Dysbindin is an established schizophrenia susceptibility gene thoroughly studied in the context of the brain. We have previously shown through a yeast two-hybrid screen that it is also a cardiac binding partner of the intercalated disc protein Myozap. Because Dysbindin is highly expressed in the heart, we aimed here at deciphering its cardiac function. Using a serum response factor (SRF) response element reporter-driven luciferase assay, we identified a robust activation of SRF signaling by Dysbindin overexpression that was associated with significant up-regulation of SRF gene targets, such as Acta1 and Actc1. Concurrently, we identified RhoA as a novel binding partner of Dysbindin. Further phenotypic and mechanistic characterization revealed that Dysbindin induced cardiac hypertrophy via RhoA-SRF and MEK1-ERK1 signaling pathways. In conclusion, we show a novel cardiac role of Dysbindin in the activation of RhoA-SRF and MEK1-ERK1 signaling pathways and in the induction of cardiac hypertrophy. Future in vivo studies should examine the significance of Dysbindin in cardiomyopathy.
Figures
Similar articles
-
TRIM24 protein promotes and TRIM32 protein inhibits cardiomyocyte hypertrophy via regulation of dysbindin protein levels.J Biol Chem. 2017 Jun 16;292(24):10180-10196. doi: 10.1074/jbc.M116.752543. Epub 2017 May 2. J Biol Chem. 2017. PMID: 28465353 Free PMC article.
-
Dysbindin deficiency Alters Cardiac BLOC-1 Complex and Myozap Levels in Mice.Cells. 2020 Oct 31;9(11):2390. doi: 10.3390/cells9112390. Cells. 2020. PMID: 33142804 Free PMC article.
-
Rho-family GTPase 1 (Rnd1) is a biomechanical stress-sensitive activator of cardiomyocyte hypertrophy.J Mol Cell Cardiol. 2019 Apr;129:130-143. doi: 10.1016/j.yjmcc.2019.01.028. Epub 2019 Feb 21. J Mol Cell Cardiol. 2019. PMID: 30797814
-
The Rac and Rho hall of fame: a decade of hypertrophic signaling hits.Circ Res. 2006 Mar 31;98(6):730-42. doi: 10.1161/01.RES.0000216039.75913.9e. Circ Res. 2006. PMID: 16574914 Review.
-
Dysbindin-containing complexes and their proposed functions in brain: from zero to (too) many in a decade.ASN Neuro. 2011 May 27;3(2):e00058. doi: 10.1042/AN20110010. ASN Neuro. 2011. PMID: 21504412 Free PMC article. Review.
Cited by
-
Fibin regulates cardiomyocyte hypertrophy and causes protein-aggregate-associated cardiomyopathy in vivo.Front Mol Biosci. 2023 Jun 5;10:1169658. doi: 10.3389/fmolb.2023.1169658. eCollection 2023. Front Mol Biosci. 2023. PMID: 37342207 Free PMC article.
-
RhoA: a dubious molecule in cardiac pathophysiology.J Biomed Sci. 2021 Apr 28;28(1):33. doi: 10.1186/s12929-021-00730-w. J Biomed Sci. 2021. PMID: 33906663 Free PMC article. Review.
-
Myozap Deficiency Promotes Adverse Cardiac Remodeling via Differential Regulation of Mitogen-activated Protein Kinase/Serum-response Factor and β-Catenin/GSK-3β Protein Signaling.J Biol Chem. 2016 Feb 19;291(8):4128-43. doi: 10.1074/jbc.M115.689620. Epub 2015 Dec 30. J Biol Chem. 2016. PMID: 26719331 Free PMC article.
-
Effect of atorvastatin on cardiomyocyte hypertrophy through suppressing MURC induced by volume overload and cyclic stretch.J Cell Mol Med. 2019 Feb;23(2):1406-1414. doi: 10.1111/jcmm.14044. Epub 2018 Dec 3. J Cell Mol Med. 2019. PMID: 30511410 Free PMC article.
-
Sumoylation-independent activation of Calcineurin-NFAT-signaling via SUMO2 mediates cardiomyocyte hypertrophy.Sci Rep. 2016 Oct 21;6:35758. doi: 10.1038/srep35758. Sci Rep. 2016. PMID: 27767176 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
