

Exposure to biomass smoke extract enhances fibronectin release from fibroblasts
- PMID: 24386310
- PMCID: PMC3873416
- DOI: 10.1371/journal.pone.0083938
Exposure to biomass smoke extract enhances fibronectin release from fibroblasts
Abstract
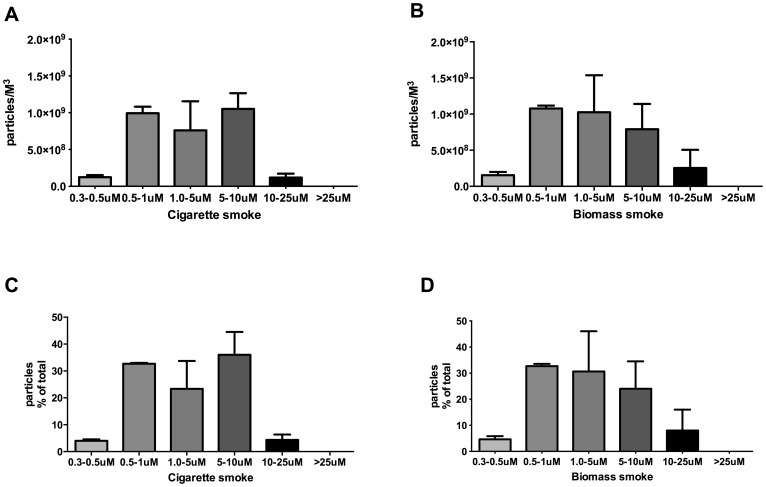
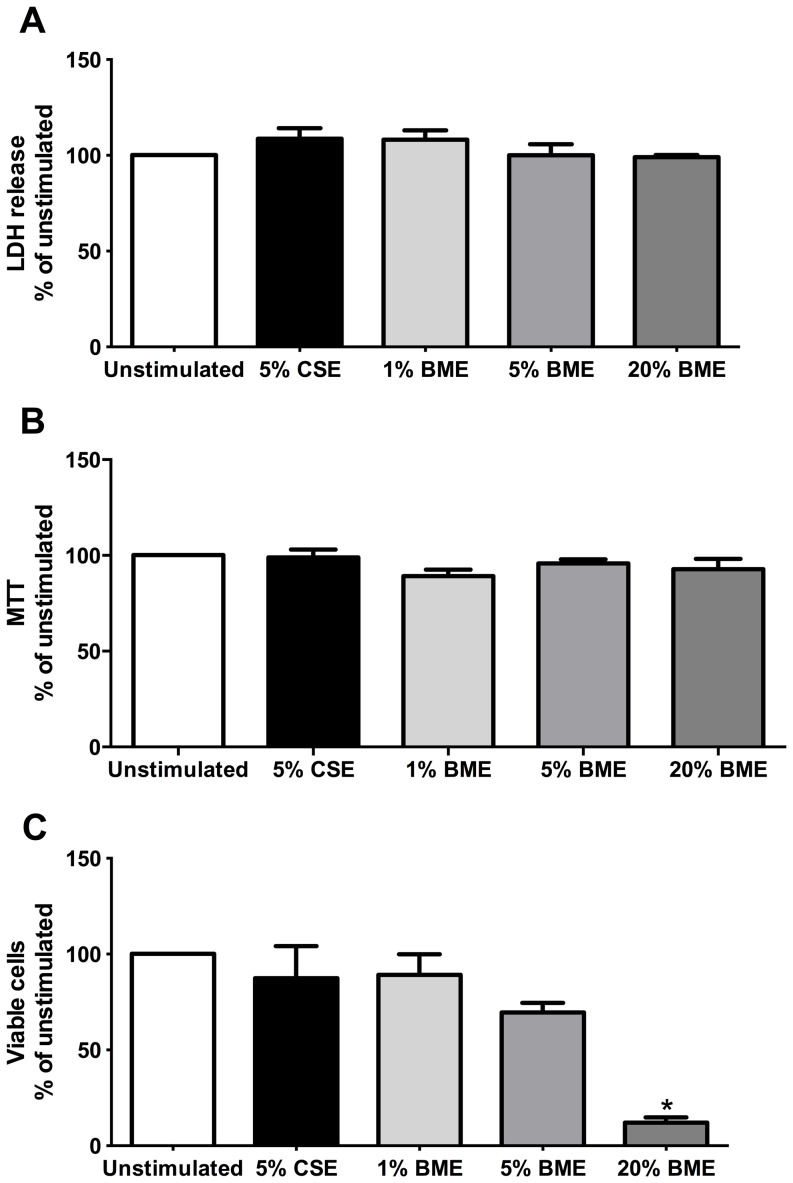
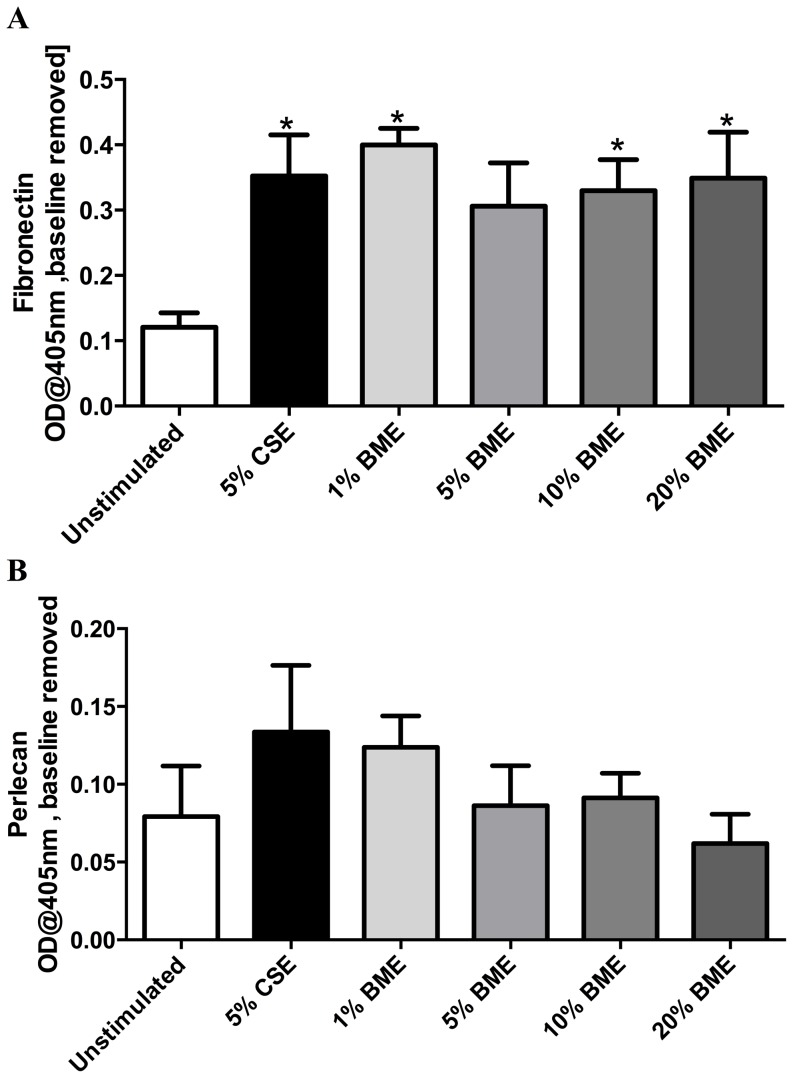
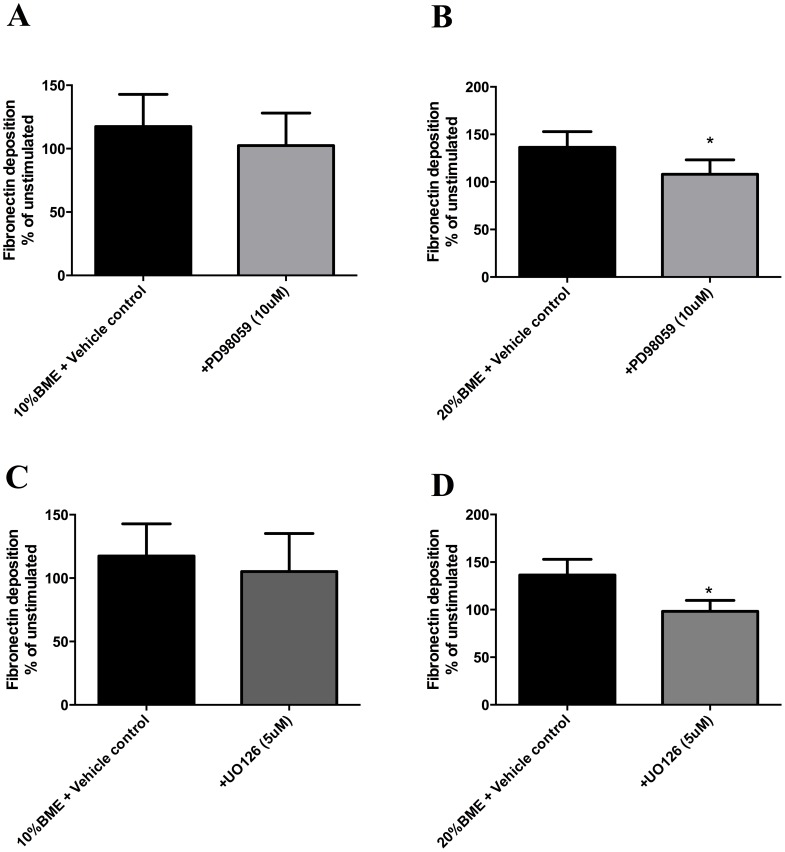
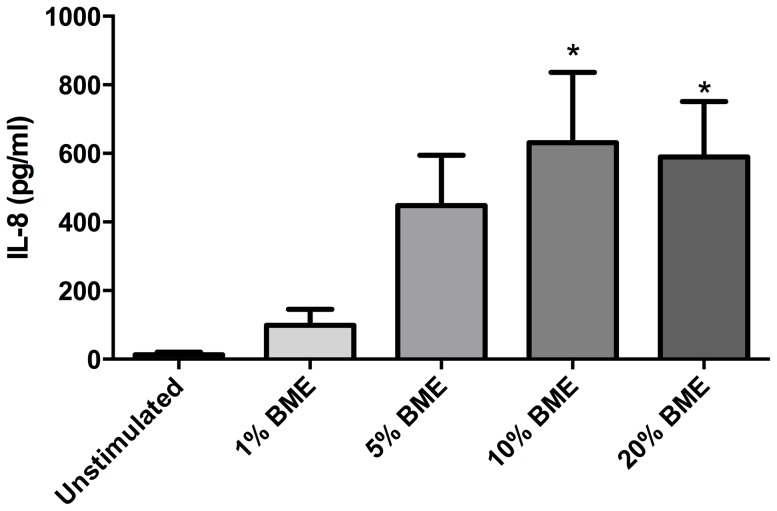
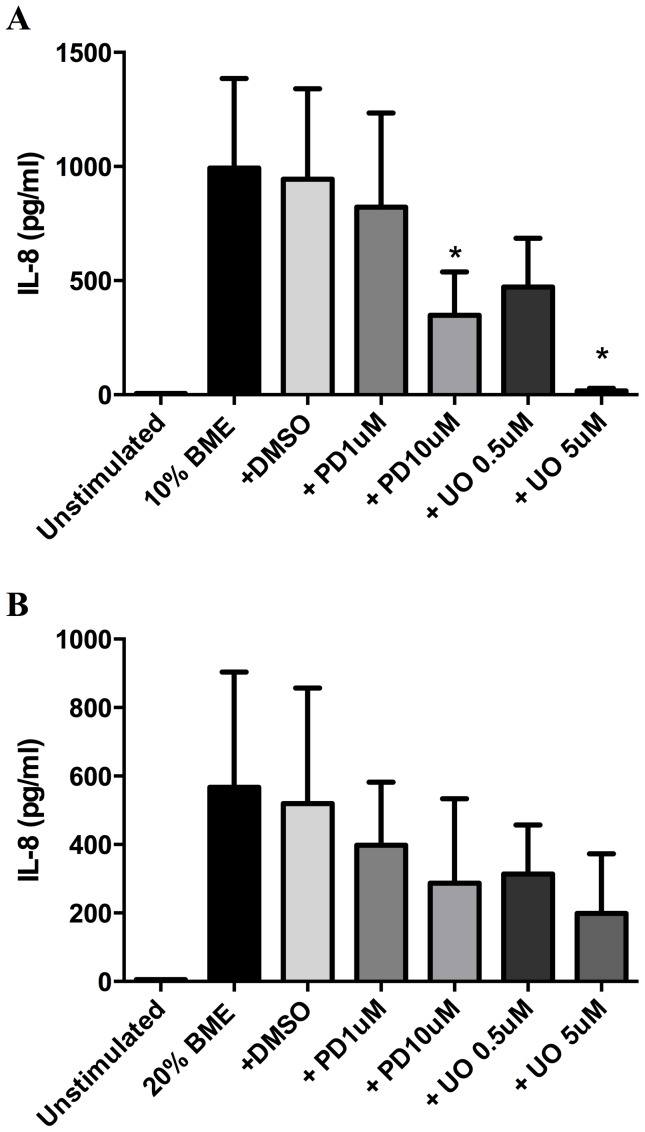
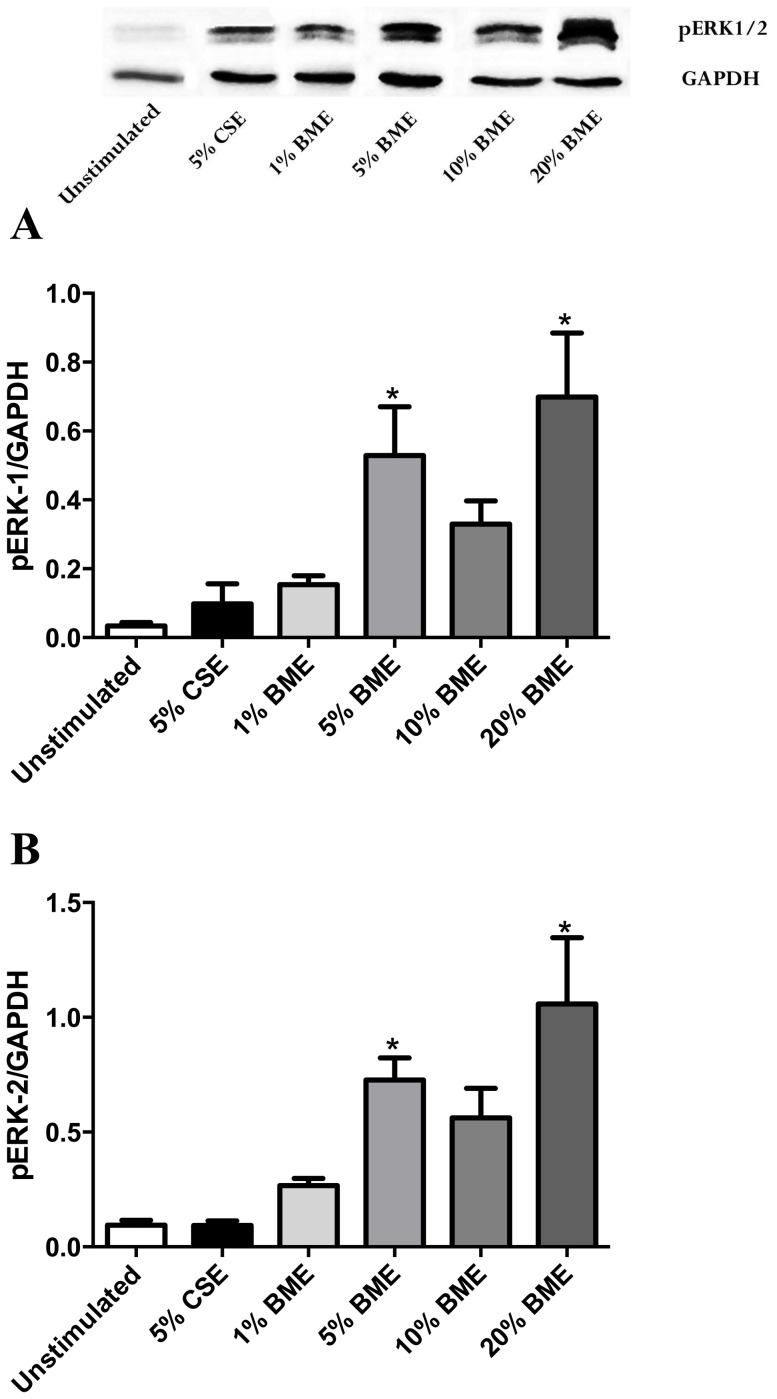
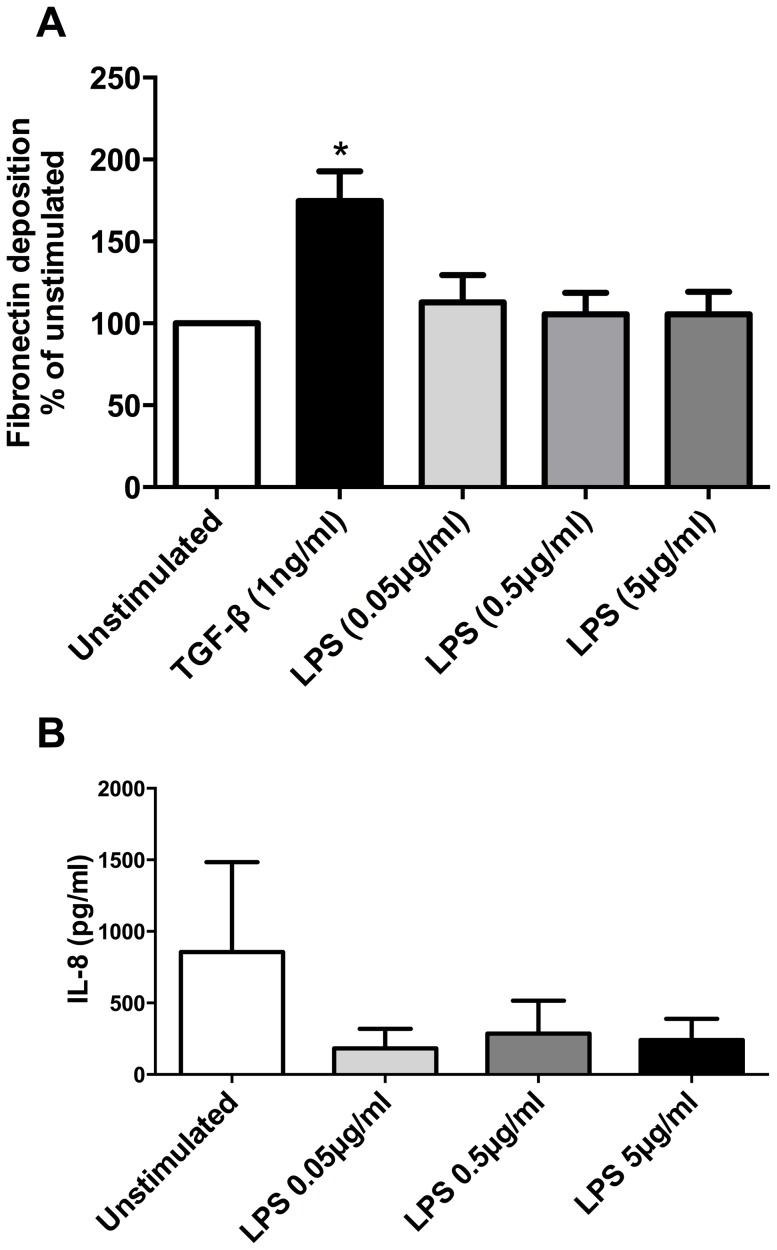
COPD induced following biomass smoke exposure has been reported to be associated with a more fibrotic phenotype than cigarette smoke induced COPD. This study aimed to investigate if biomass smoke induced extracellular matrix (ECM) protein production from primary human lung fibroblasts in vitro. Primary human lung fibroblasts (n=5-10) were stimulated in vitro for up to 72 hours with increasing concentrations of biomass smoke extract (BME) or cigarette smoke extract (CSE) prior to being assessed for deposition of ECM proteins, cytokine release, and activation of intracellular signalling molecules. Deposition of the ECM proteins perlecan and fibronectin was upregulated by both CSE (p<0.05) and BME (p<0.05). The release of the neutrophilic chemokine IL-8 was also enhanced by BME. ERK1/2 phosphorylation was significantly upregulated by BME (p<0.05). Chemical inhibition of ERK signalling molecules partially attenuated these effects (p<0.05). Stimulation with endotoxin had no effect. This study demonstrated that BME had similar effects to CSE in vitro and had the capacity to directly induce fibrosis by upregulating production of ECM proteins. The mechanisms by which both biomass and cigarette smoke exposure cause lung damage may be similar.
Conflict of interest statement
Figures
Similar articles
-
Matrix proteins from smoke-exposed fibroblasts are pro-proliferative.Am J Respir Cell Mol Biol. 2012 Jan;46(1):34-9. doi: 10.1165/rcmb.2010-0426OC. Am J Respir Cell Mol Biol. 2012. PMID: 21778414
-
Caspase inhibition restores collagen Iα1 and fibronectin release in cigarette smoke extract-exposed human lung fibroblasts.Am J Physiol Lung Cell Mol Physiol. 2025 Feb 1;328(2):L239-L252. doi: 10.1152/ajplung.00214.2024. Epub 2025 Jan 8. Am J Physiol Lung Cell Mol Physiol. 2025. PMID: 39772929
-
Biomass Smoke Exposure Enhances Rhinovirus-Induced Inflammation in Primary Lung Fibroblasts.Int J Mol Sci. 2016 Aug 25;17(9):1403. doi: 10.3390/ijms17091403. Int J Mol Sci. 2016. PMID: 27571064 Free PMC article.
-
alpha,beta-Unsaturated aldehydes contained in cigarette smoke elicit IL-8 release in pulmonary cells through mitogen-activated protein kinases.Am J Physiol Lung Cell Mol Physiol. 2009 May;296(5):L839-48. doi: 10.1152/ajplung.90570.2008. Epub 2009 Mar 13. Am J Physiol Lung Cell Mol Physiol. 2009. PMID: 19286926
-
Smad3 mediates cigarette smoke extract (CSE) induction of VEGF release by human fetal lung fibroblasts.Toxicol Lett. 2013 Jul 4;220(2):126-34. doi: 10.1016/j.toxlet.2013.04.011. Epub 2013 Apr 22. Toxicol Lett. 2013. PMID: 23618901
Cited by
-
Fisetin Mitigates Chronic Lung Injury Induced by Benzo(a)Pyrene by Regulation of Inflammation and Oxidative Stress.Curr Issues Mol Biol. 2025 Mar 19;47(3):209. doi: 10.3390/cimb47030209. Curr Issues Mol Biol. 2025. PMID: 40136463 Free PMC article.
-
Evidence of Biomass Smoke Exposure as a Causative Factor for the Development of COPD.Toxics. 2017 Dec 1;5(4):36. doi: 10.3390/toxics5040036. Toxics. 2017. PMID: 29194400 Free PMC article. Review.
-
Implementation of the Care Bundle for the Management of Chronic Obstructive Pulmonary Disease with/without Heart Failure.J Clin Med. 2024 Mar 12;13(6):1621. doi: 10.3390/jcm13061621. J Clin Med. 2024. PMID: 38541845 Free PMC article. Review.
-
Air pollution and cardiovascular disease: Can the Australian bushfires and global COVID-19 pandemic of 2020 convince us to change our ways?Bioessays. 2021 Sep;43(9):e2100046. doi: 10.1002/bies.202100046. Epub 2021 Jun 9. Bioessays. 2021. PMID: 34106476 Free PMC article. Review.
-
Development of Inhalable Spray Dried Nitrofurantoin Formulations for the Treatment of Emphysema.Pharmaceutics. 2022 Dec 31;15(1):146. doi: 10.3390/pharmaceutics15010146. Pharmaceutics. 2022. PMID: 36678775 Free PMC article.
References
-
- Salvi SS, Barnes PJ (2009) Chronic obstructive pulmonary disease in non-smokers. Lancet 374: 733–743. - PubMed
-
- Salvi S, Barnes PJ (2010) Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest 138: 3–6. - PubMed
-
- Desalu OO, Adekoya AO, Ampitan BA (2010) Increased risk of respiratory symptoms and chronic bronchitis in women using biomass fuels in Nigeria. J Bras Pneumol 36: 441–446. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
