

Characterizing the infection-induced transcriptome of Nasonia vitripennis reveals a preponderance of taxonomically-restricted immune genes
- PMID: 24386321
- PMCID: PMC3873987
- DOI: 10.1371/journal.pone.0083984
Characterizing the infection-induced transcriptome of Nasonia vitripennis reveals a preponderance of taxonomically-restricted immune genes
Abstract
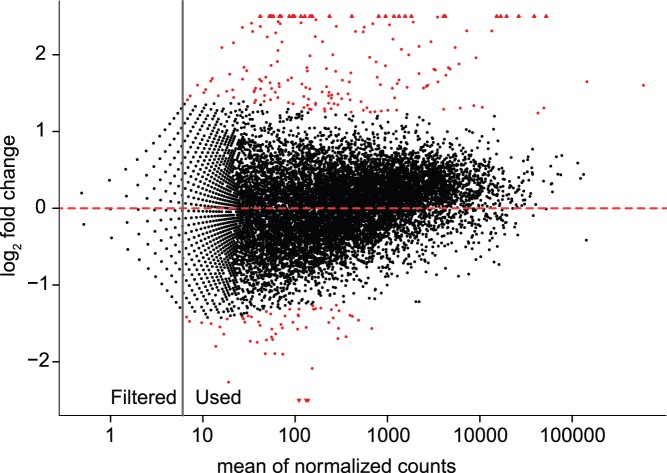
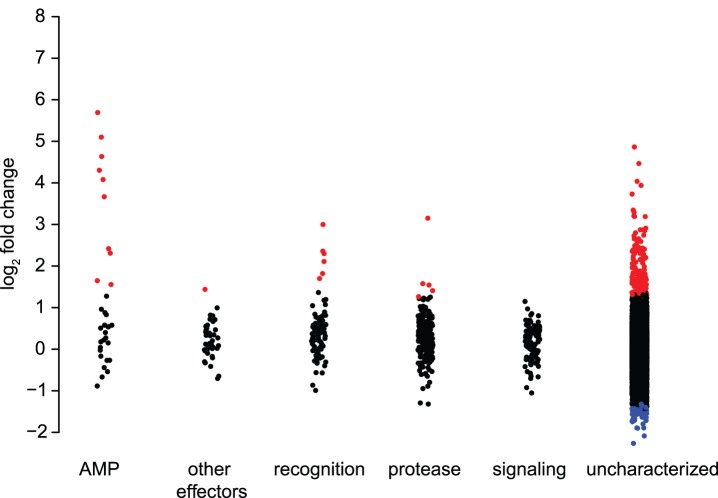
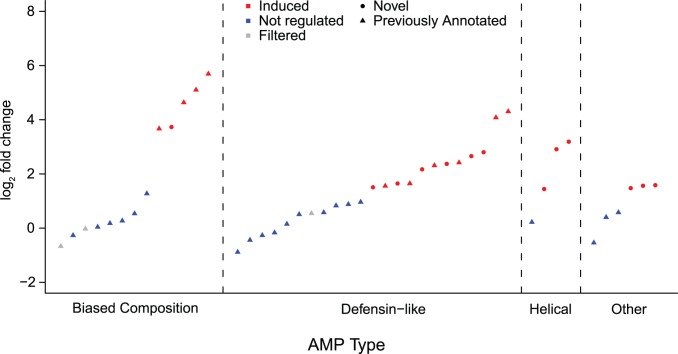
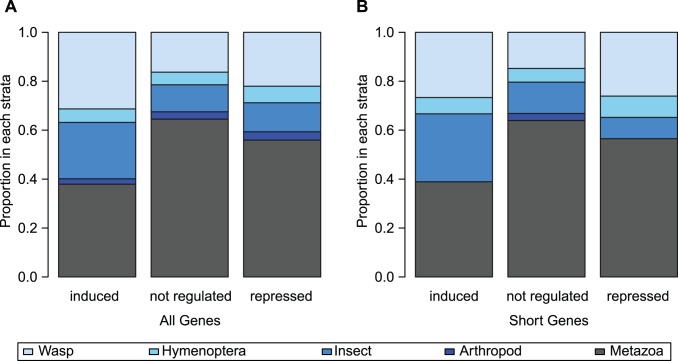
The innate immune system in insects consists of a conserved core signaling network and rapidly diversifying effector and recognition components, often containing a high proportion of taxonomically-restricted genes. In the absence of functional annotation, genes encoding immune system proteins can thus be difficult to identify, as homology-based approaches generally cannot detect lineage-specific genes. Here, we use RNA-seq to compare the uninfected and infection-induced transcriptome in the parasitoid wasp Nasonia vitripennis to identify genes regulated by infection. We identify 183 genes significantly up-regulated by infection and 61 genes significantly down-regulated by infection. We also produce a new homology-based immune catalog in N. vitripennis, and show that most infection-induced genes cannot be assigned an immune function from homology alone, suggesting the potential for substantial novel immune components in less well-studied systems. Finally, we show that a high proportion of these novel induced genes are taxonomically restricted, highlighting the rapid evolution of immune gene content. The combination of functional annotation using RNA-seq and homology-based annotation provides a robust method to characterize the innate immune response across a wide variety of insects, and reveals significant novel features of the Nasonia immune response.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
