

A de novo convergence of autism genetics and molecular neuroscience
- PMID: 24387789
- PMCID: PMC4077788
- DOI: 10.1016/j.tins.2013.11.005
A de novo convergence of autism genetics and molecular neuroscience
Abstract
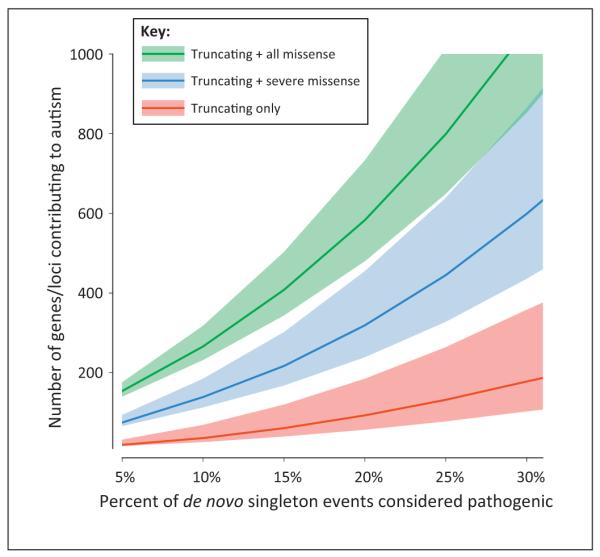
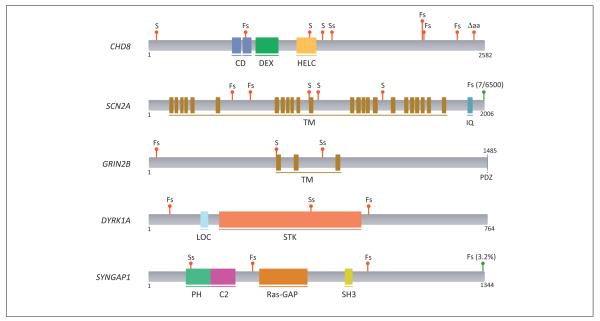
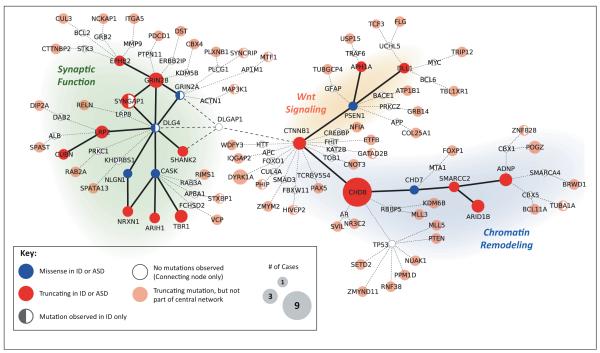
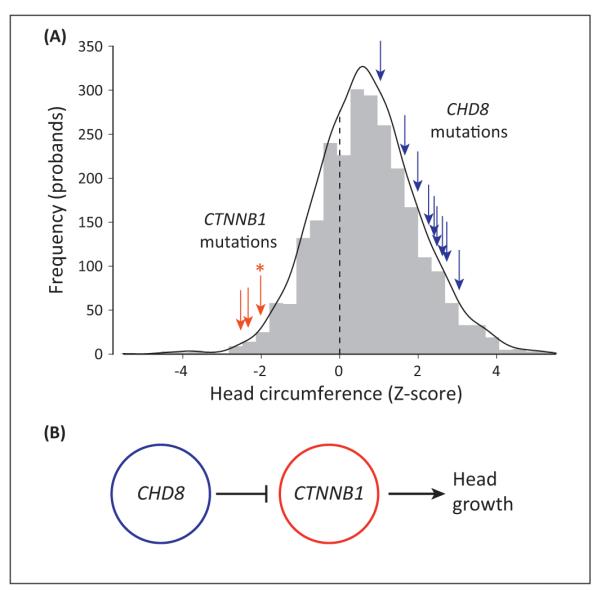
Autism spectrum disorder (ASD) and intellectual disability (ID) are neurodevelopmental disorders with large genetic components, but identification of pathogenic genes has proceeded slowly because hundreds of loci are involved. New exome sequencing technology has identified novel rare variants and has found that sporadic cases of ASD/ID are enriched for disruptive de novo mutations. Targeted large-scale resequencing studies have confirmed the significance of specific loci, including chromodomain helicase DNA binding protein 8 (CHD8), sodium channel, voltage-gated, type II, alpha subunit (SCN2A), dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), and catenin (cadherin-associated protein), beta 1, 88 kDa (CTNNB1, beta-catenin). We review recent studies and suggest that they have led to a convergence on three functional pathways: (i) chromatin remodeling; (ii) wnt signaling during development; and (iii) synaptic function. These pathways and genes significantly expand the neurobiological targets for study, and suggest a path for future genetic and functional studies.
Keywords: autism genetics; autism spectrum disorder; copy number variant; exome sequencing; intellectual disability; single nucleotide variant.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
References
-
- Fu Y-H, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. - PubMed
-
- Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. - PubMed
-
- Matsuura T, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997;15:74–77. - PubMed
-
- Steffenburg S, et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child Psychol. Psychiatry. 1989;30:405–416. - PubMed
-
- Bailey A, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 1995;25:63–77. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
