

Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia
- PMID: 24388663
- PMCID: PMC3928657
- DOI: 10.1016/j.ajhg.2013.12.005
Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia
Abstract
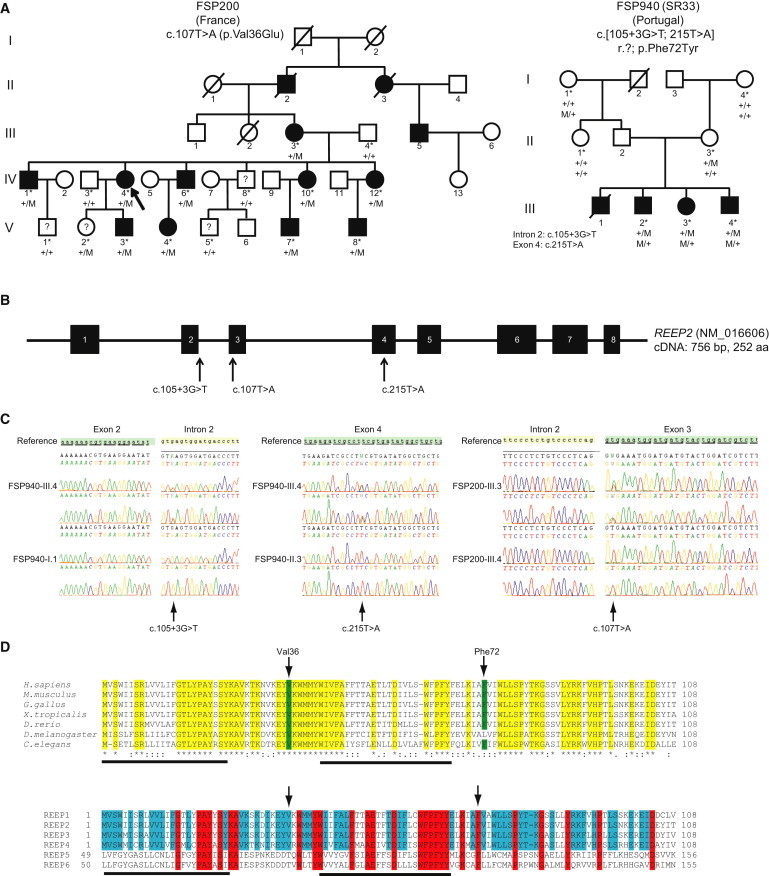
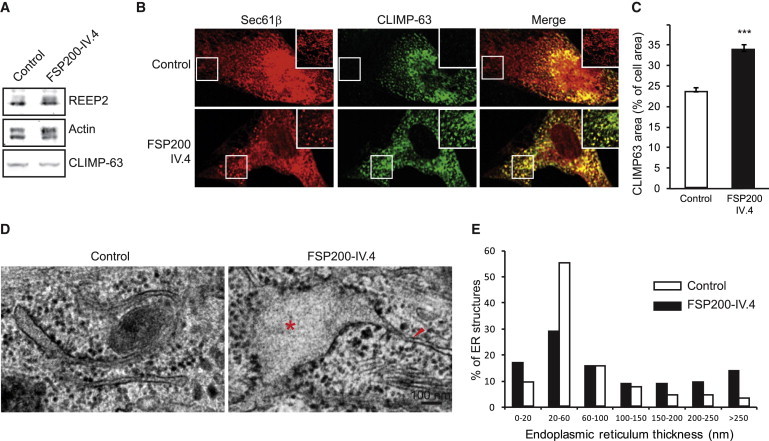
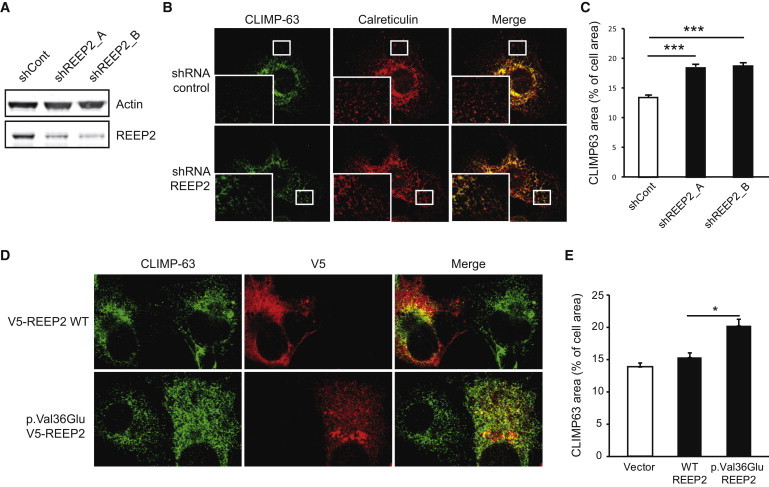
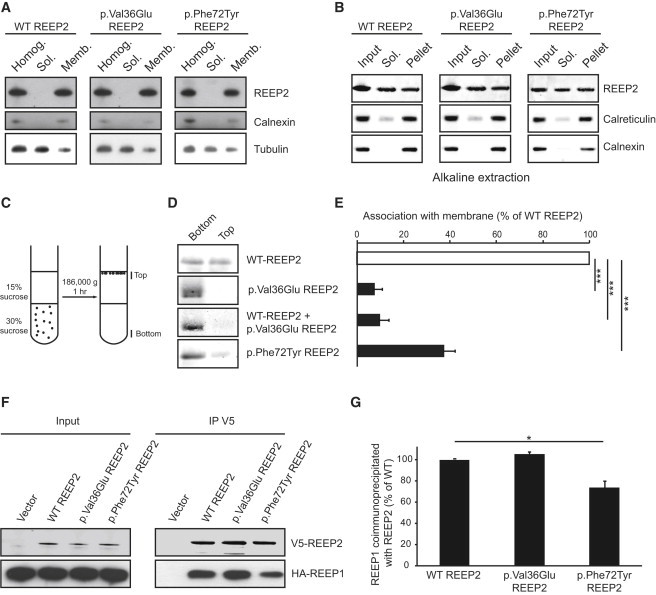
Hereditary spastic paraplegias (HSPs) are clinically and genetically heterogeneous neurological conditions. Their main pathogenic mechanisms are thought to involve alterations in endomembrane trafficking, mitochondrial function, and lipid metabolism. With a combination of whole-genome mapping and exome sequencing, we identified three mutations in REEP2 in two families with HSP: a missense variant (c.107T>A [p.Val36Glu]) that segregated in the heterozygous state in a family with autosomal-dominant inheritance and a missense change (c.215T>A [p.Phe72Tyr]) that segregated in trans with a splice site mutation (c.105+3G>T) in a family with autosomal-recessive transmission. REEP2 belongs to a family of proteins that shape the endoplasmic reticulum, an organelle that was altered in fibroblasts from an affected subject. In vitro, the p.Val36Glu variant in the autosomal-dominant family had a dominant-negative effect; it inhibited the normal binding of wild-type REEP2 to membranes. The missense substitution p.Phe72Tyr, in the recessive family, decreased the affinity of the mutant protein for membranes that, together with the splice site mutation, is expected to cause complete loss of REEP2 function. Our findings illustrate how dominant and recessive inheritance can be explained by the effects and nature of mutations in the same gene. They have also important implications for genetic diagnosis and counseling in clinical practice because of the association of various modes of inheritance to this new clinico-genetic entity.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
A Nepalese family with an REEP2 mutation: clinical and genetic study.J Hum Genet. 2021 Jul;66(7):749-752. doi: 10.1038/s10038-020-00882-x. Epub 2021 Feb 1. J Hum Genet. 2021. PMID: 33526816
-
A novel heterozygous variant in ERLIN2 causes autosomal dominant pure hereditary spastic paraplegia.Eur J Neurol. 2018 Jul;25(7):943-e71. doi: 10.1111/ene.13625. Epub 2018 Apr 15. Eur J Neurol. 2018. PMID: 29528531
-
Evidence for autosomal recessive inheritance in SPG3A caused by homozygosity for a novel ATL1 missense mutation.Eur J Hum Genet. 2014 Oct;22(10):1180-4. doi: 10.1038/ejhg.2014.5. Epub 2014 Jan 29. Eur J Hum Genet. 2014. PMID: 24473461 Free PMC article.
-
More autosomal dominant SPG18 cases than recessive? The first AD-SPG18 pedigree in Chinese and literature review.Brain Behav. 2021 Dec;11(12):e32395. doi: 10.1002/brb3.2395. Epub 2021 Nov 3. Brain Behav. 2021. PMID: 34734492 Free PMC article. Review.
-
Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms.Exp Neurol. 2014 Nov;261:518-39. doi: 10.1016/j.expneurol.2014.06.011. Epub 2014 Jun 20. Exp Neurol. 2014. PMID: 24954637 Review.
Cited by
-
Loss of spatacsin impairs cholesterol trafficking and calcium homeostasis.Commun Biol. 2019 Oct 17;2:380. doi: 10.1038/s42003-019-0615-z. eCollection 2019. Commun Biol. 2019. PMID: 31637311 Free PMC article.
-
NeurodegenERation: The Central Role for ER Contacts in Neuronal Function and Axonopathy, Lessons From Hereditary Spastic Paraplegias and Related Diseases.Front Neurosci. 2019 Oct 11;13:1051. doi: 10.3389/fnins.2019.01051. eCollection 2019. Front Neurosci. 2019. PMID: 31680803 Free PMC article. Review.
-
BmREEPa Is a Novel Gene that Facilitates BmNPV Entry into Silkworm Cells.PLoS One. 2015 Dec 14;10(12):e0144575. doi: 10.1371/journal.pone.0144575. eCollection 2015. PLoS One. 2015. PMID: 26656276 Free PMC article.
-
Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa.Am J Hum Genet. 2016 Dec 1;99(6):1305-1315. doi: 10.1016/j.ajhg.2016.10.008. Epub 2016 Nov 23. Am J Hum Genet. 2016. PMID: 27889058 Free PMC article.
-
Challenges and Controversies in the Genetic Diagnosis of Hereditary Spastic Paraplegia.Curr Neurol Neurosci Rep. 2021 Feb 28;21(4):15. doi: 10.1007/s11910-021-01099-x. Curr Neurol Neurosci Rep. 2021. PMID: 33646413 Free PMC article. Review.
References
-
- Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. - PubMed
-
- Tallaksen C.M., Dürr A., Brice A. Recent advances in hereditary spastic paraplegia. Curr. Opin. Neurol. 2001;14:457–463. - PubMed
-
- Stevanin G., Ruberg M., Brice A. Recent advances in the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2008;8:198–210. - PubMed
-
- Coutinho P., Barros J., Zemmouri R., Guimarães J., Alves C., Chorão R., Lourenço E., Ribeiro P., Loureiro J.L., Santos J.V. Clinical heterogeneity of autosomal recessive spastic paraplegias: analysis of 106 patients in 46 families. Arch. Neurol. 1999;56:943–949. - PubMed
-
- Erichsen A.K., Koht J., Stray-Pedersen A., Abdelnoor M., Tallaksen C.M. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain. 2009;132:1577–1588. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
