

Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico
- PMID: 24390345
- PMCID: PMC4127086
- DOI: 10.1038/nature12828
Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico
Abstract
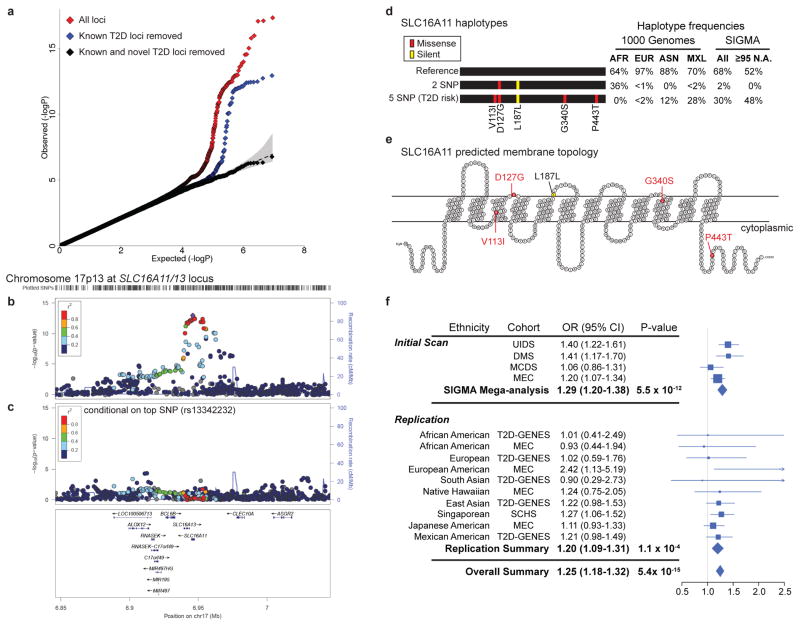
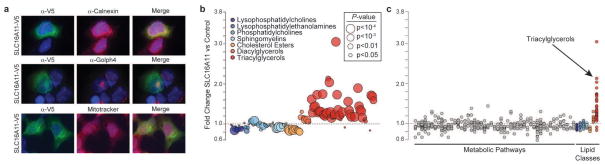
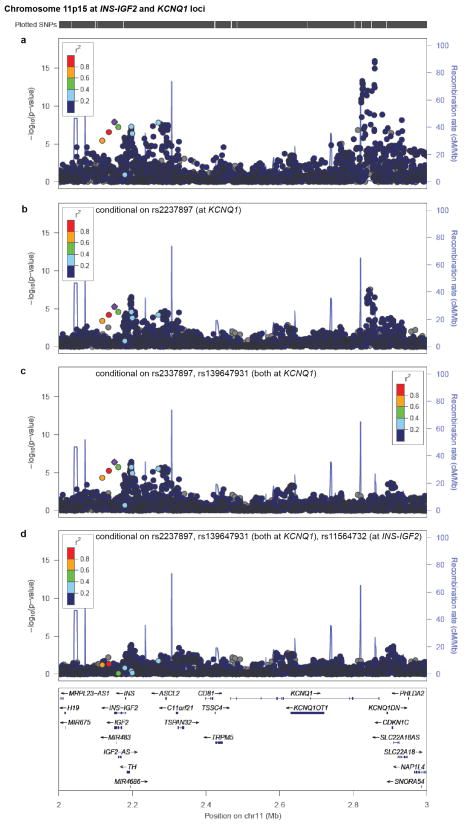
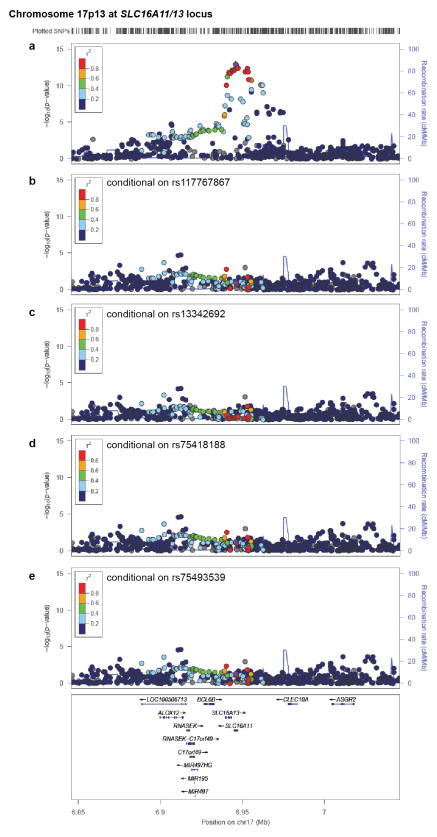
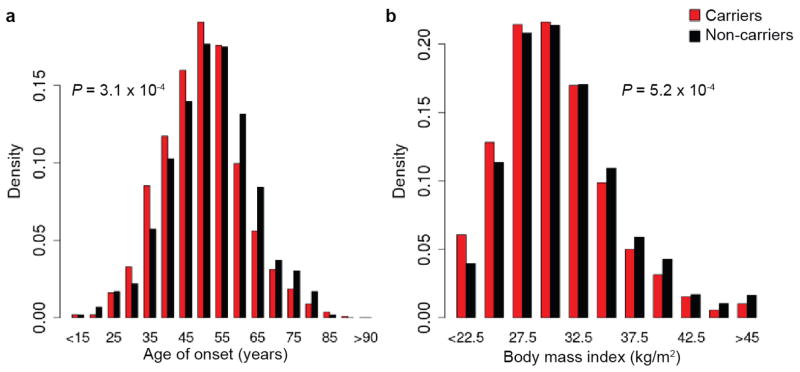
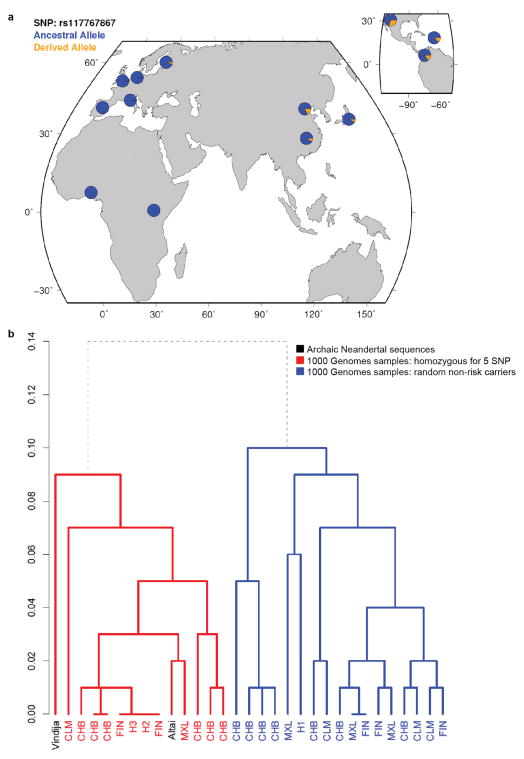
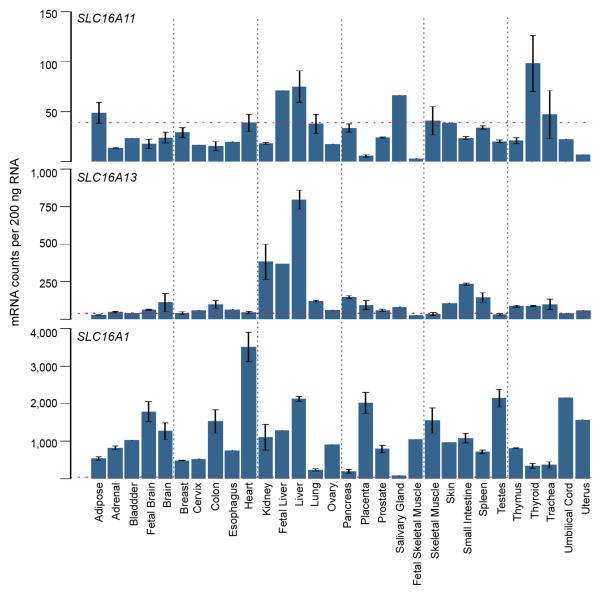
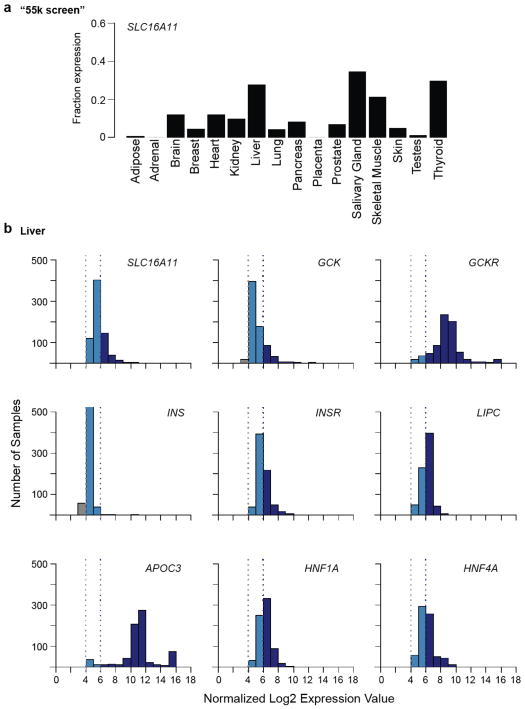
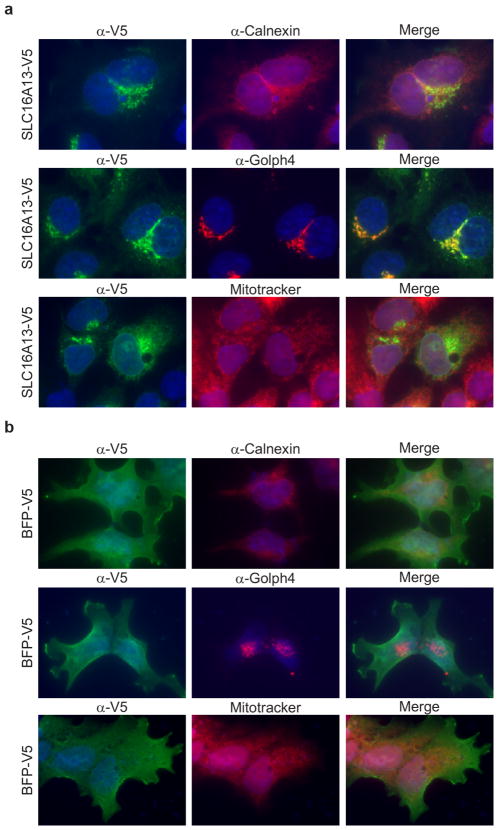
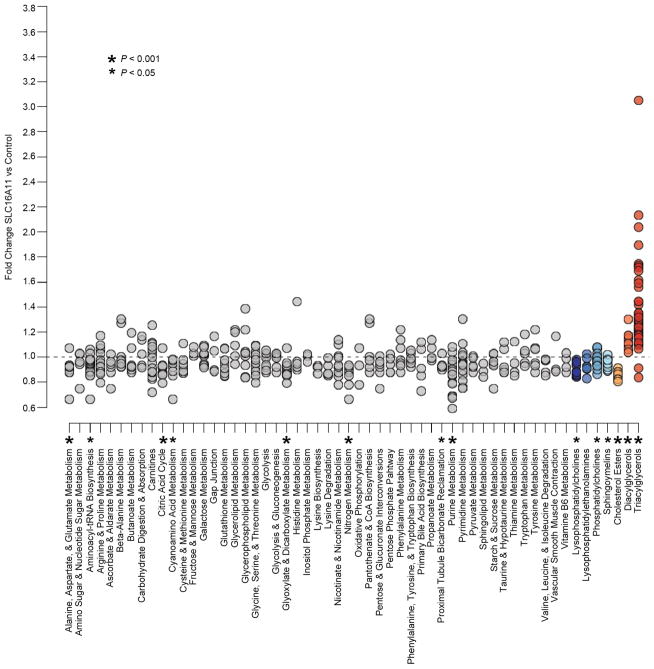
Performing genetic studies in multiple human populations can identify disease risk alleles that are common in one population but rare in others, with the potential to illuminate pathophysiology, health disparities, and the population genetic origins of disease alleles. Here we analysed 9.2 million single nucleotide polymorphisms (SNPs) in each of 8,214 Mexicans and other Latin Americans: 3,848 with type 2 diabetes and 4,366 non-diabetic controls. In addition to replicating previous findings, we identified a novel locus associated with type 2 diabetes at genome-wide significance spanning the solute carriers SLC16A11 and SLC16A13 (P = 3.9 × 10(-13); odds ratio (OR) = 1.29). The association was stronger in younger, leaner people with type 2 diabetes, and replicated in independent samples (P = 1.1 × 10(-4); OR = 1.20). The risk haplotype carries four amino acid substitutions, all in SLC16A11; it is present at ~50% frequency in Native American samples and ~10% in east Asian, but is rare in European and African samples. Analysis of an archaic genome sequence indicated that the risk haplotype introgressed into modern humans via admixture with Neanderthals. The SLC16A11 messenger RNA is expressed in liver, and V5-tagged SLC16A11 protein localizes to the endoplasmic reticulum. Expression of SLC16A11 in heterologous cells alters lipid metabolism, most notably causing an increase in intracellular triacylglycerol levels. Despite type 2 diabetes having been well studied by genome-wide association studies in other populations, analysis in Mexican and Latin American individuals identified SLC16A11 as a novel candidate gene for type 2 diabetes with a possible role in triacylglycerol metabolism.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Epidemiology: Genetic T2DM risk factor found.Nat Rev Endocrinol. 2014 Mar;10(3):128. doi: 10.1038/nrendo.2013.273. Epub 2014 Jan 14. Nat Rev Endocrinol. 2014. PMID: 24419356 No abstract available.
References
-
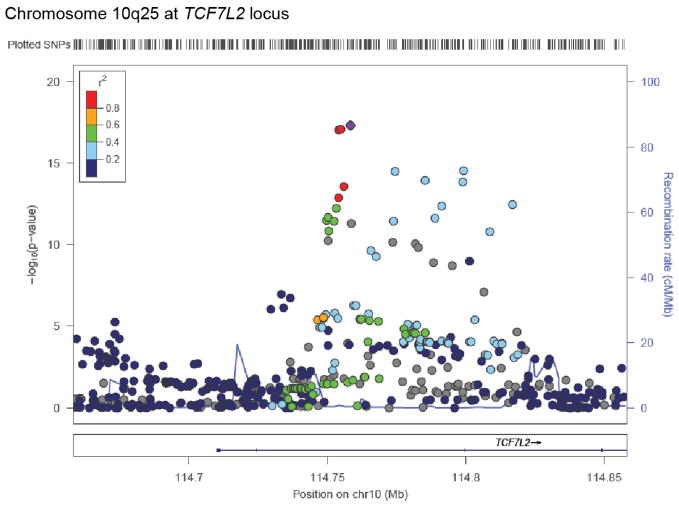
- Grant SFA, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature genetics. 2006;38:320–323. doi: http://www.nature.com/ng/journal/v38/n3/suppinfo/ng1732_S1.html. - PubMed
-
- Unoki H, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nature genetics. 2008;40:1098–1102. doi: http://www.nature.com/ng/journal/v40/n9/suppinfo/ng.208_S1.html. - PubMed
-
- Yasuda K, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nature genetics. 2008;40:1092–1097. doi: http://www.nature.com/ng/journal/v40/n9/suppinfo/ng.207_S1.html. - PubMed
-
- Villalpando S, et al. Prevalence and distribution of type 2 diabetes mellitus in Mexican adult population: a probabilistic survey. Salud Pública de México. 2010;52:S19–S26. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- UM1 CA164973/CA/NCI NIH HHS/United States
- R01 CA80205/CA/NCI NIH HHS/United States
- U01 DK057295/DK/NIDDK NIH HHS/United States
- U01DK085526/DK/NIDDK NIH HHS/United States
- F32 HG005944/HG/NHGRI NIH HHS/United States
- R01 DK047482/DK/NIDDK NIH HHS/United States
- R01 CA063464/CA/NCI NIH HHS/United States
- R01HL24799/HL/NHLBI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 DK053889/DK/NIDDK NIH HHS/United States
- R01DK053889/DK/NIDDK NIH HHS/United States
- R01 DK057295/DK/NIDDK NIH HHS/United States
- R01 CA144034/CA/NCI NIH HHS/United States
- R01 HG006399/HG/NHGRI NIH HHS/United States
- R01 DK042273/DK/NIDDK NIH HHS/United States
- R35 CA053890/CA/NCI NIH HHS/United States
- U01 DK085526/DK/NIDDK NIH HHS/United States
- P01 HL045522/HL/NHLBI NIH HHS/United States
- R01 CA054281/CA/NCI NIH HHS/United States
- U01 CA063464/CA/NCI NIH HHS/United States
- U01 CA164973/CA/NCI NIH HHS/United States
- R37 CA054281/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
