

Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology
- PMID: 24391846
- PMCID: PMC3877102
- DOI: 10.1371/journal.pone.0083922
Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology
Abstract
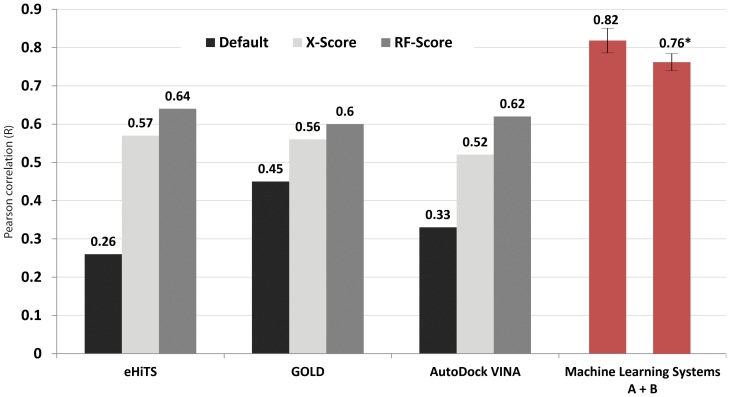
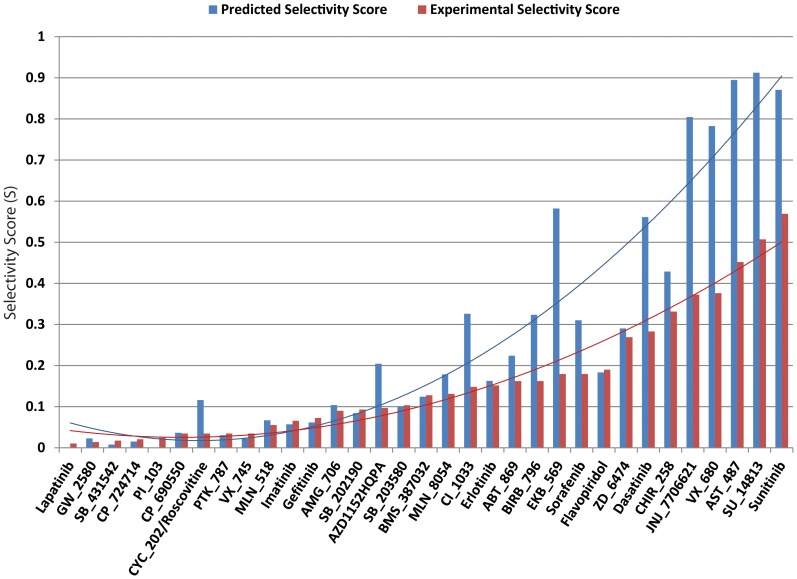
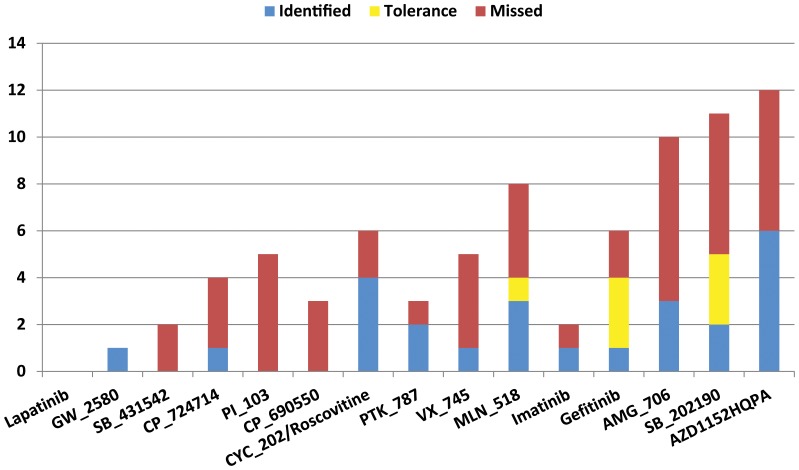
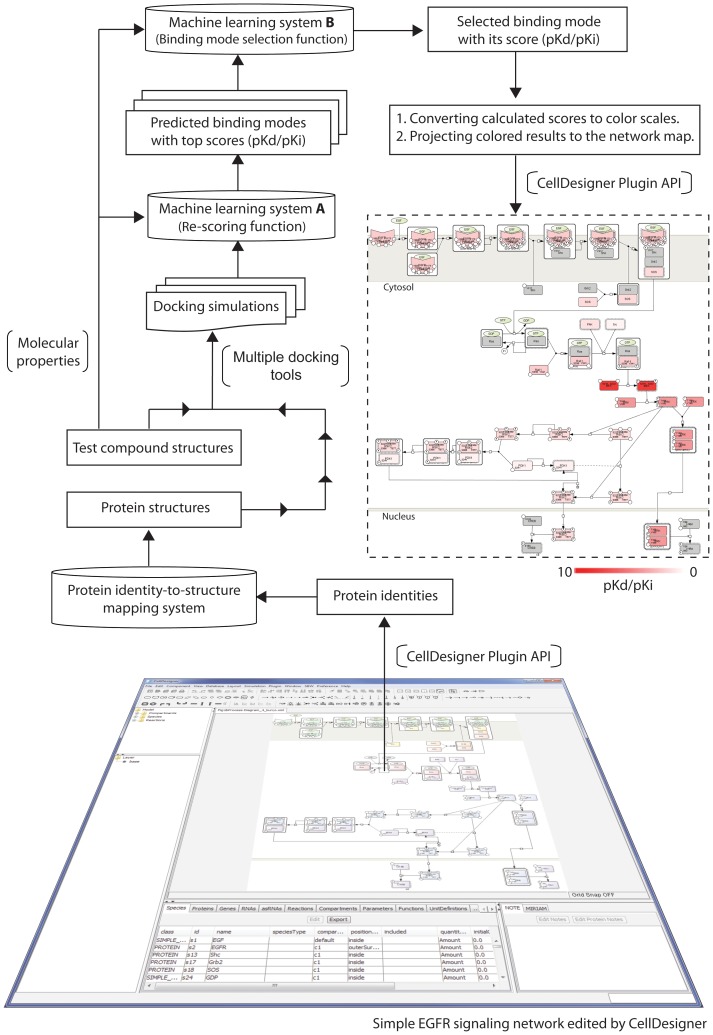
Increased availability of bioinformatics resources is creating opportunities for the application of network pharmacology to predict drug effects and toxicity resulting from multi-target interactions. Here we present a high-precision computational prediction approach that combines two elaborately built machine learning systems and multiple molecular docking tools to assess binding potentials of a test compound against proteins involved in a complex molecular network. One of the two machine learning systems is a re-scoring function to evaluate binding modes generated by docking tools. The second is a binding mode selection function to identify the most predictive binding mode. Results from a series of benchmark validations and a case study show that this approach surpasses the prediction reliability of other techniques and that it also identifies either primary or off-targets of kinase inhibitors. Integrating this approach with molecular network maps makes it possible to address drug safety issues by comprehensively investigating network-dependent effects of a drug or drug candidate.
Conflict of interest statement
Figures
References
-
- Force T, Krause DS, Van Etten RA (2007) Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer 7: 332–344. - PubMed
-
- Cases M, Mestres J (2009) A chemogenomic approach to drug discovery: focus on cardiovascular diseases. Drug Discov Today 14: 479–485. - PubMed
-
- Csermely P, Agoston V, Pongor S (2005) The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci 26: 178–182. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
