

Turning off AKT: PHLPP as a drug target
- PMID: 24392697
- PMCID: PMC4082184
- DOI: 10.1146/annurev-pharmtox-011112-140338
Turning off AKT: PHLPP as a drug target
Abstract
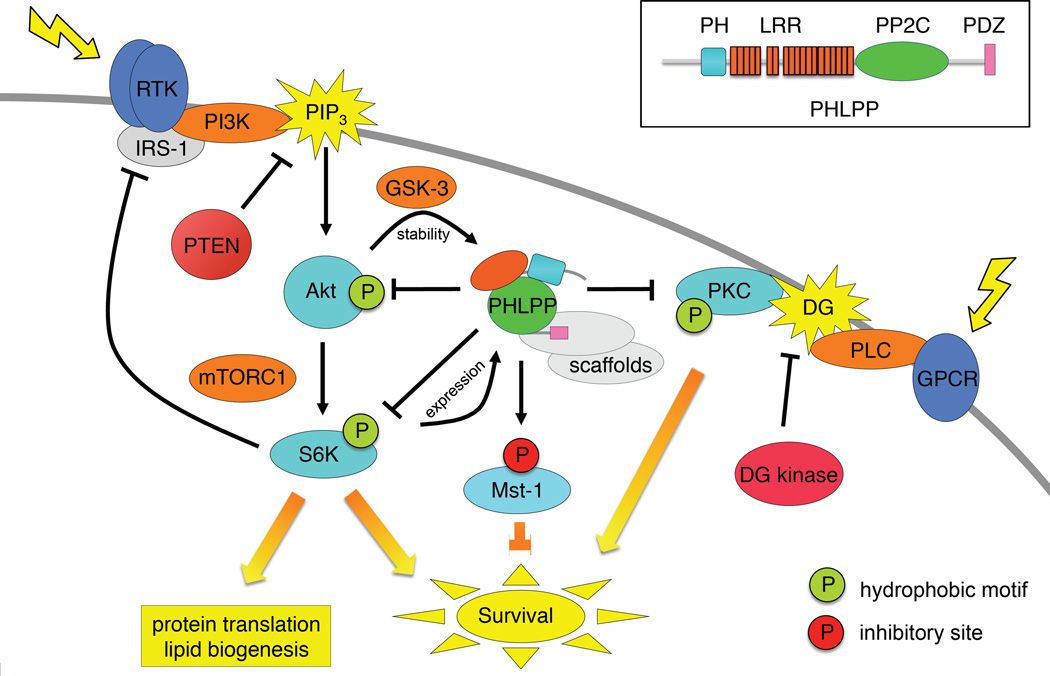
Precise control of the balance between protein phosphorylation, catalyzed by protein kinases, and protein dephosphorylation, catalyzed by protein phosphatases, is essential for cellular homeostasis. Dysregulation of this balance leads to pathophysiological states, driving diseases such as cancer, heart disease, and diabetes. Aberrant phosphorylation of components of the pathways that control cell growth and cell survival are particularly prevalent in cancer. One of the most studied tumor suppressors in these pathways is the lipid phosphatase PTEN (phosphatase and tensin homolog deleted on chromosome ten), which dephosphorylates the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3), thus preventing activation of the oncogenic kinase AKT (v-akt murine thymoma viral oncogene homolog). In 2005, the discovery of a family of protein phosphatases whose members directly dephosphorylate and inactivate AKT introduced a new negative regulator of the phosphoinositide 3-kinase (PI3K) oncogenic pathway. Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) isozymes comprise a novel tumor suppressor family whose two members, PHLPP1 and PHLPP2, are deleted as frequently as PTEN in cancers such as those of the prostate. PHLPP is thus a novel therapeutic target to suppress oncogenic pathways and is a potential candidate biomarker to stratify patients for the appropriate targeted therapeutics. This review discusses the role of PHLPP in terminating AKT signaling and how pharmacological intervention would impact this pathway.
Figures



References
-
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. - PubMed
-
- Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57:652–658. - PubMed
-
- Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
