

PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast
- PMID: 24393166
- PMCID: PMC4053934
- DOI: 10.1186/gb-2014-15-1-r8
PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast
Abstract
Background: Nrd1 and Nab3 are essential sequence-specific yeast RNA binding proteins that function as a heterodimer in the processing and degradation of diverse classes of RNAs. These proteins also regulate several mRNA coding genes; however, it remains unclear exactly what percentage of the mRNA component of the transcriptome these proteins control. To address this question, we used the pyCRAC software package developed in our laboratory to analyze CRAC and PAR-CLIP data for Nrd1-Nab3-RNA interactions.
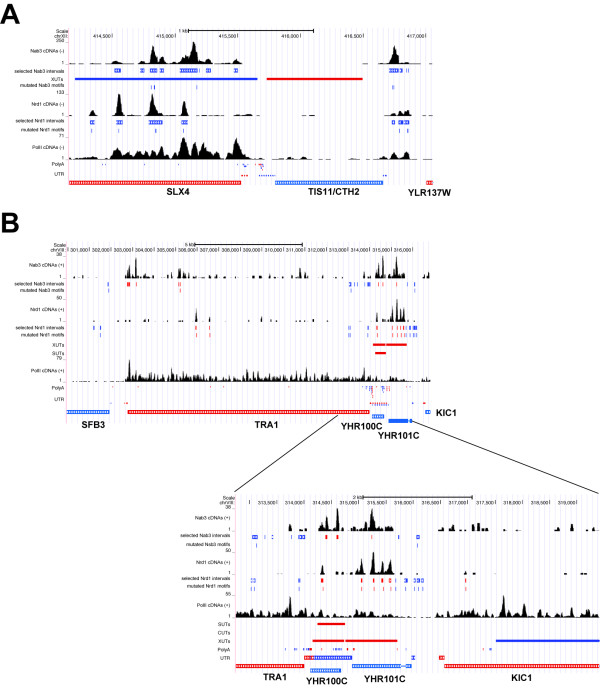
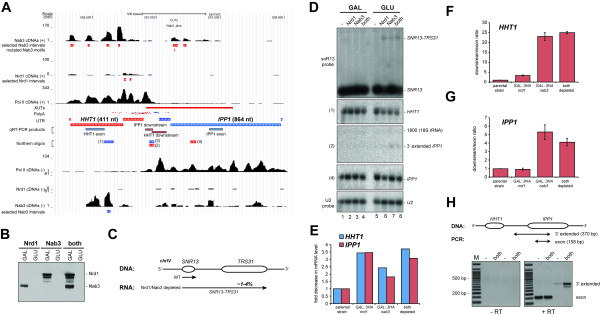
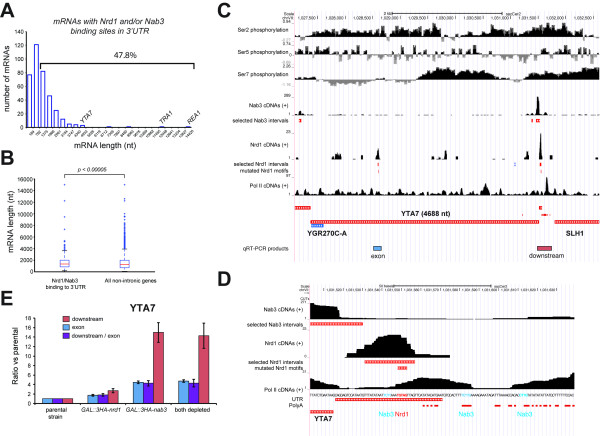
Results: We generated high-resolution maps of Nrd1-Nab3-RNA interactions, from which we have uncovered hundreds of new Nrd1-Nab3 mRNA targets, representing between 20 and 30% of protein-coding transcripts. Although Nrd1 and Nab3 showed a preference for binding near 5' ends of relatively short transcripts, they bound transcripts throughout coding sequences and 3' UTRs. Moreover, our data for Nrd1-Nab3 binding to 3' UTRs was consistent with a role for these proteins in the termination of transcription. Our data also support a tight integration of Nrd1-Nab3 with the nutrient response pathway. Finally, we provide experimental evidence for some of our predictions, using northern blot and RT-PCR assays.
Conclusions: Collectively, our data support the notion that Nrd1 and Nab3 function is tightly integrated with the nutrient response and indicate a role for these proteins in the regulation of many mRNA coding genes. Further, we provide evidence to support the hypothesis that Nrd1-Nab3 represents a failsafe termination mechanism in instances of readthrough transcription.
Figures
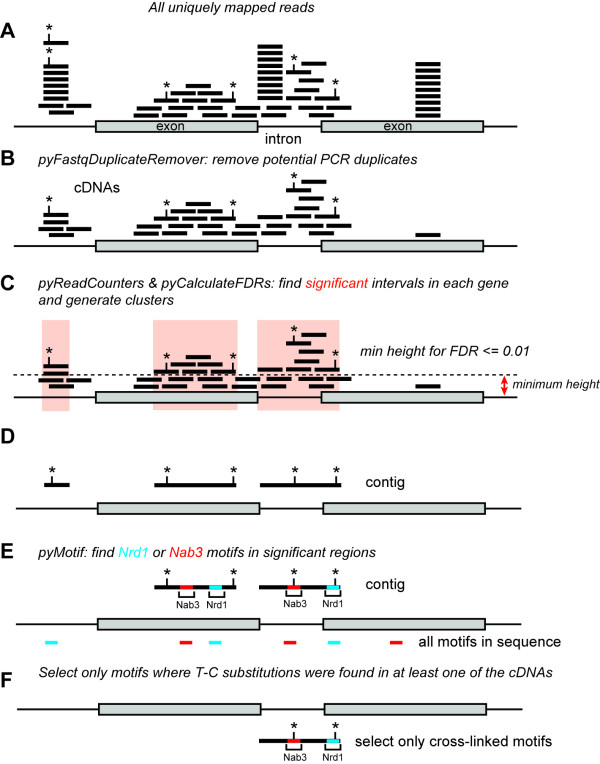
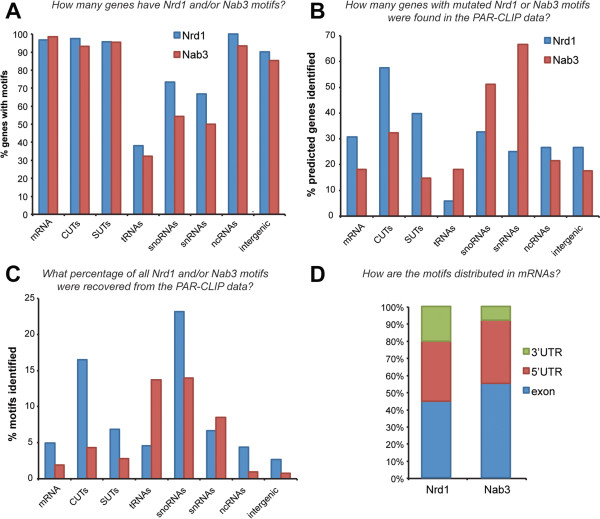
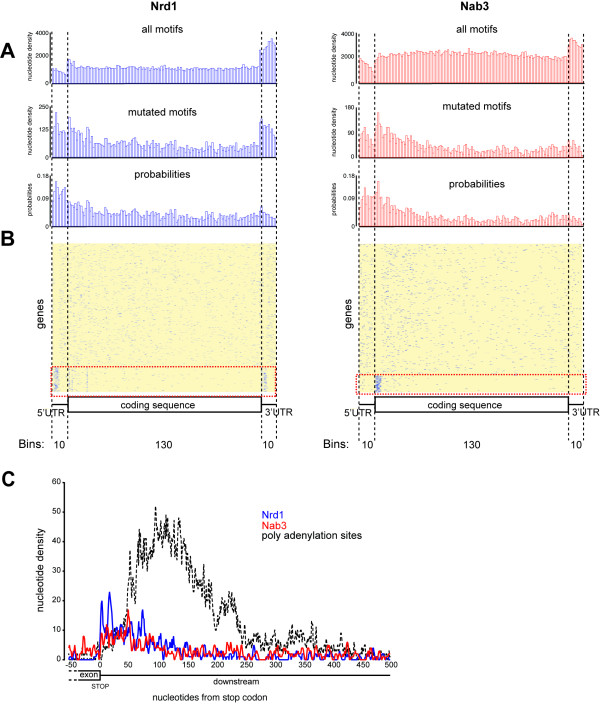

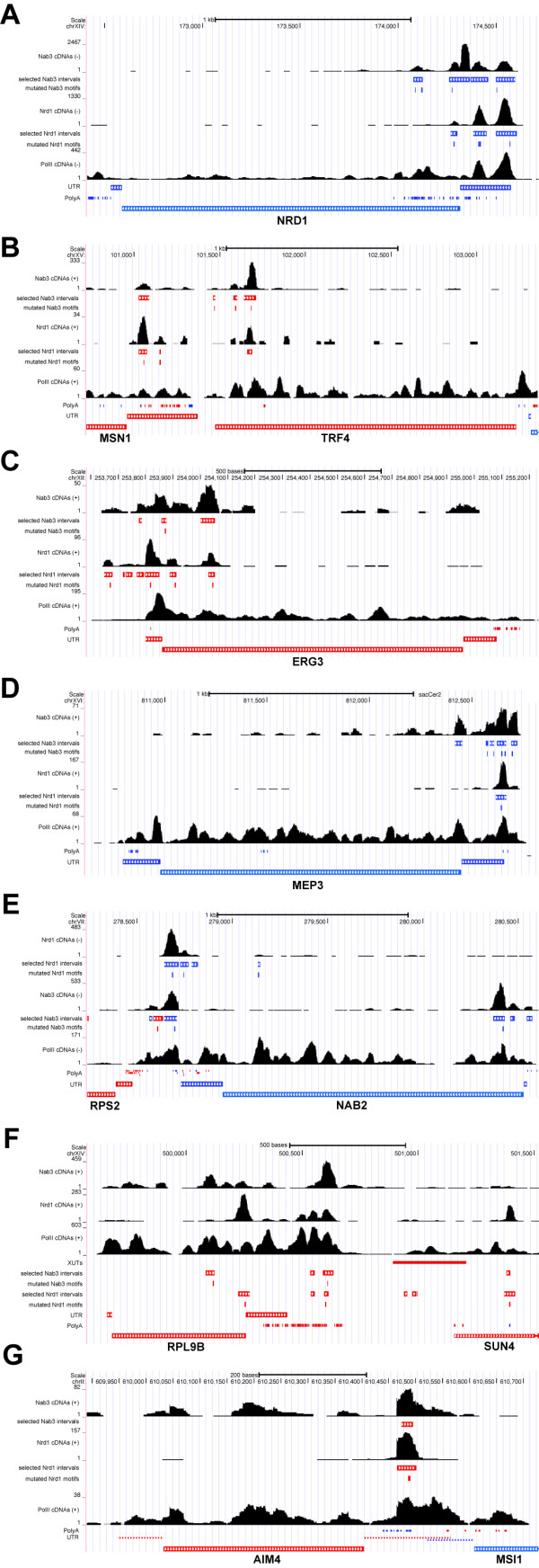
References
-
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
