

Examining weak protein-protein interactions in start codon recognition via NMR spectroscopy
- PMID: 24393460
- PMCID: PMC3991732
- DOI: 10.1111/febs.12667
Examining weak protein-protein interactions in start codon recognition via NMR spectroscopy
Abstract
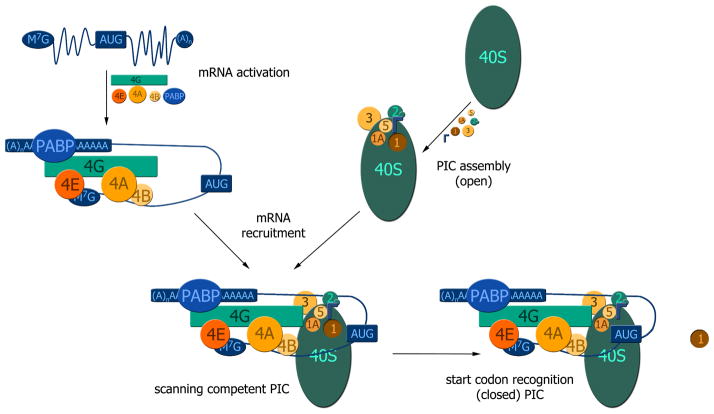
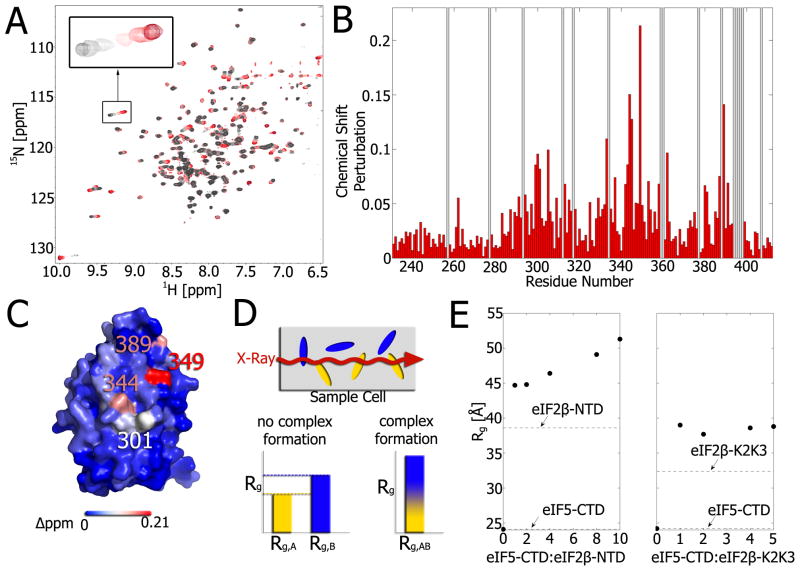
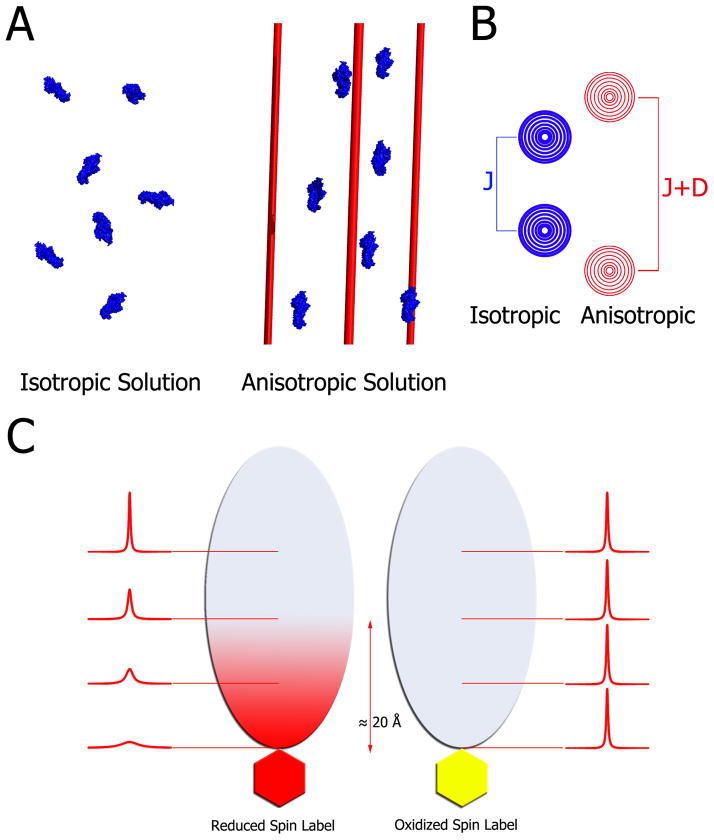
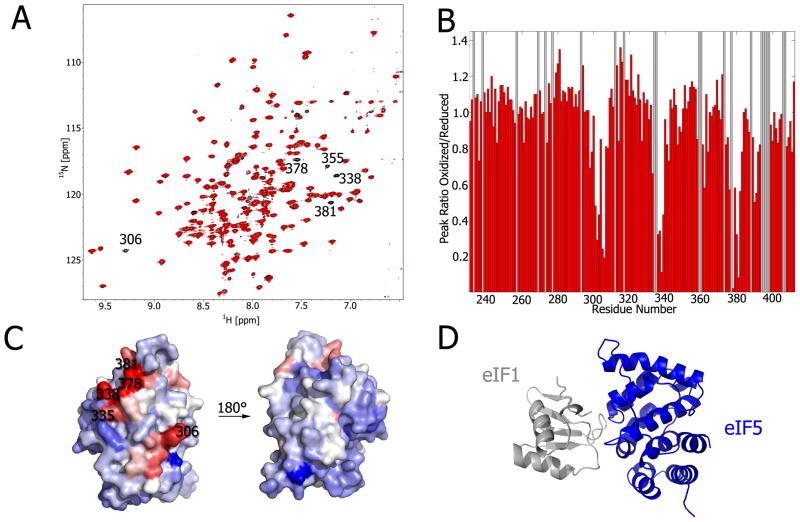
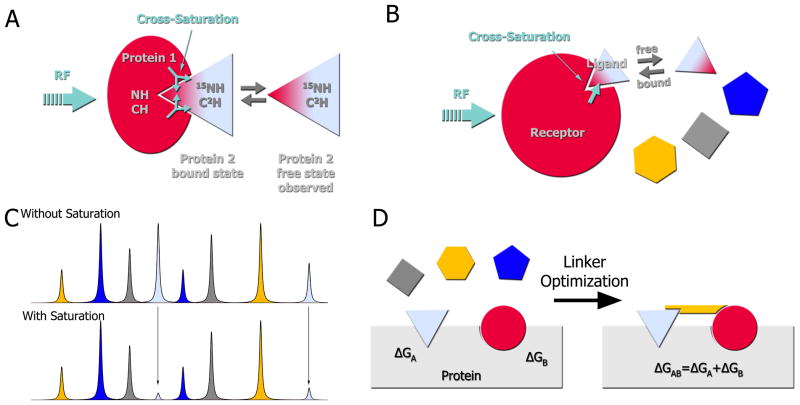
Weak protein-protein interactions are critical in numerous biological processes. Unfortunately, they are difficult to characterize due to the high concentrations required for the production and detection of the complex population. The inherent sensitivity of NMR spectroscopy to the chemical environment makes it an excellent tool to tackle this problem. NMR permits the exploration of interactions over a range of affinities, yielding essential insights into dynamic biological processes. The conversion of messanger RNA to protein is one such process that requires the coordinated association of many low-affinity proteins. During start codon recognition, eukaryotic initiation factors assemble into high-order complexes that bind messanger RNA and bring it to the ribosome for decoding. Many of the structures of the eukaryotic initiation factors have been determined; however, little is known regarding the weak binary complexes formed and their structure-function mechanisms. Herein, we use start codon recognition as a model system to review the relevant NMR methods for the characterization of weak interactions and the development of small molecule inhibitors.
Keywords: chemical shift perturbation; cross saturation; fragment based screening; initiation factors; nuclear magnetic resonance; paramagnetic relaxation enhancement; residual dipolar couplings; small angle X-Ray scattering reconstitution assay; translation initiation; weak interactions.
© 2013 FEBS.
Figures
Similar articles
-
Characterizing weak protein-protein complexes by NMR residual dipolar couplings.Eur Biophys J. 2011 Dec;40(12):1371-81. doi: 10.1007/s00249-011-0720-5. Epub 2011 Jun 28. Eur Biophys J. 2011. PMID: 21710303 Review.
-
Communication between eukaryotic translation initiation factors 5 and 1A within the ribosomal pre-initiation complex plays a role in start site selection.J Mol Biol. 2006 Feb 24;356(3):724-37. doi: 10.1016/j.jmb.2005.11.083. Epub 2005 Dec 15. J Mol Biol. 2006. PMID: 16380131
-
Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation.RNA. 2009 Jan;15(1):138-52. doi: 10.1261/rna.1318509. Epub 2008 Nov 24. RNA. 2009. PMID: 19029312 Free PMC article.
-
eIF1 Loop 2 interactions with Met-tRNAi control the accuracy of start codon selection by the scanning preinitiation complex.Proc Natl Acad Sci U S A. 2018 May 1;115(18):E4159-E4168. doi: 10.1073/pnas.1800938115. Epub 2018 Apr 16. Proc Natl Acad Sci U S A. 2018. PMID: 29666249 Free PMC article.
-
Should I stay or should I go? Eukaryotic translation initiation factors 1 and 1A control start codon recognition.J Biol Chem. 2008 Oct 10;283(41):27345-27349. doi: 10.1074/jbc.R800031200. Epub 2008 Jun 30. J Biol Chem. 2008. PMID: 18593708 Free PMC article. Review.
Cited by
-
Weak protein complexes: challenging to study but essential for life.FEBS J. 2014 Apr;281(8):1948-9. doi: 10.1111/febs.12744. FEBS J. 2014. PMID: 24860873 Free PMC article. Review. No abstract available.
-
Towards interpretation of intermolecular paramagnetic relaxation enhancement outside the fast exchange limit.J Biomol NMR. 2016 Sep;66(1):1-7. doi: 10.1007/s10858-016-0053-x. Epub 2016 Aug 24. J Biomol NMR. 2016. PMID: 27558624
-
Endogenous modulators of neurotrophin signaling: Landscape of the transient ATP-NGF interactions.Comput Struct Biotechnol J. 2021 May 7;19:2938-2949. doi: 10.1016/j.csbj.2021.05.009. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34136093 Free PMC article.
-
Driving force of biomolecular liquid-liquid phase separation probed by nuclear magnetic resonance spectroscopy.Biophys Rep. 2022 Apr 30;8(2):90-99. doi: 10.52601/bpr.2022.210034. Biophys Rep. 2022. PMID: 37287829 Free PMC article.
-
The molecular mechanism of nuclear transport revealed by atomic-scale measurements.Elife. 2015 Sep 15;4:e10027. doi: 10.7554/eLife.10027. Elife. 2015. PMID: 26371551 Free PMC article.
References
-
- Ben-Shem A, Jenner L, Yusupova G, Yusupov M. Crystal structure of the eukaryotic ribosome. Science. 2010;330:1203–1209. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
