

Selective vulnerability to neurodegenerative disease: the curious case of Prion Protein
- PMID: 24396151
- PMCID: PMC3882045
- DOI: 10.1242/dmm.012146
Selective vulnerability to neurodegenerative disease: the curious case of Prion Protein
Abstract
The mechanisms underlying the selective targeting of specific brain regions by different neurodegenerative diseases is one of the most intriguing mysteries in medicine. For example, it is known that Alzheimer's disease primarily affects parts of the brain that play a role in memory, whereas Parkinson's disease predominantly affects parts of the brain that are involved in body movement. However, the reasons that other brain regions remain unaffected in these diseases are unknown. A better understanding of the phenomenon of selective vulnerability is required for the development of targeted therapeutic approaches that specifically protect affected neurons, thereby altering the disease course and preventing its progression. Prion diseases are a fascinating group of neurodegenerative diseases because they exhibit a wide phenotypic spectrum caused by different sequence perturbations in a single protein. The possible ways that mutations affecting this protein can cause several distinct neurodegenerative diseases are explored in this Review to highlight the complexity underlying selective vulnerability. The premise of this article is that selective vulnerability is determined by the interaction of specific protein conformers and region-specific microenvironments harboring unique combinations of subcellular components such as metals, chaperones and protein translation machinery. Given the abundance of potential contributory factors in the neurodegenerative process, a better understanding of how these factors interact will provide invaluable insight into disease mechanisms to guide therapeutic discovery.
Keywords: Huntington’s disease; Knock-in mice; Neurodegeneration; Neuropathology; Prion diseases; Spinocerebellar ataxia.
Figures
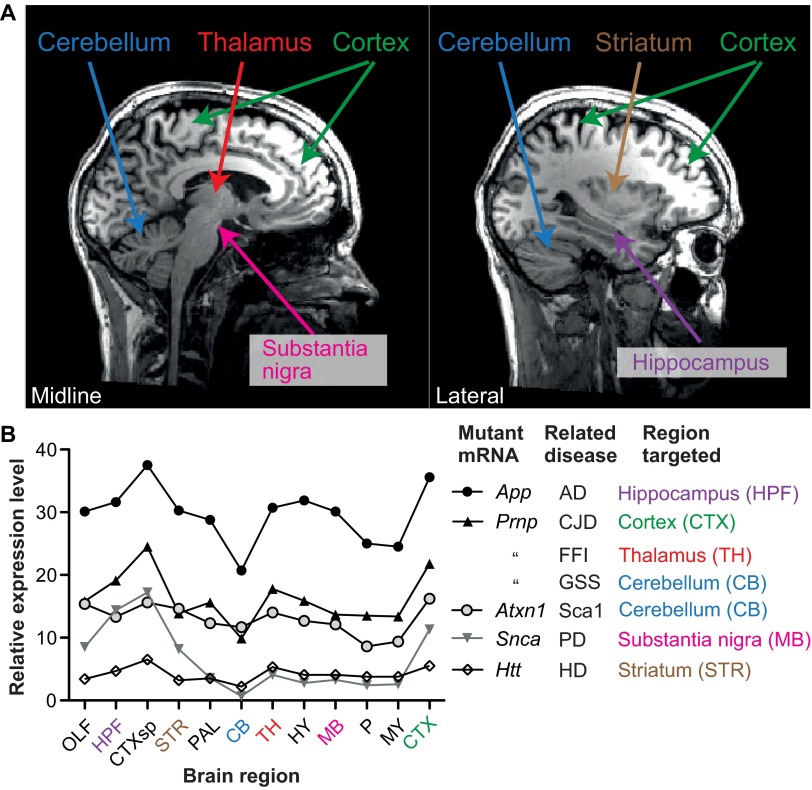
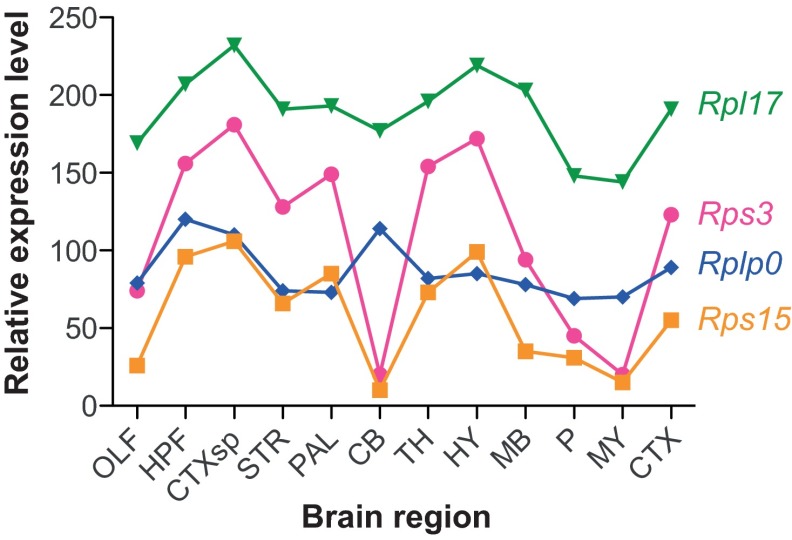
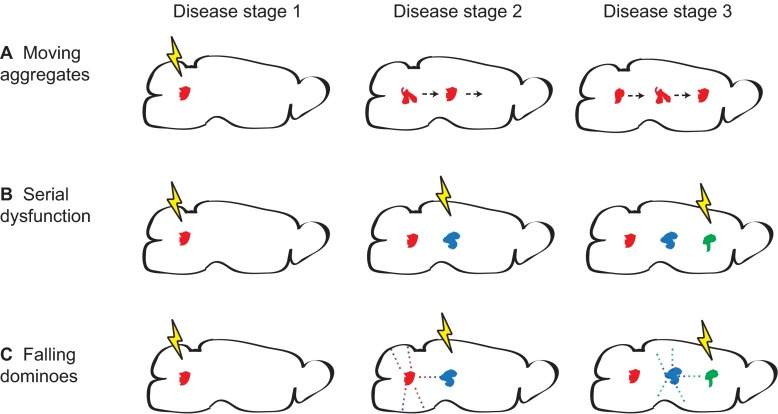
References
-
- Aguzzi A., Rajendran L. (2009). The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790 - PubMed
-
- Aidt F. H., Hasholt L. F., Christiansen M., Laursen H. (2013). Localization of A11-reactive oligomeric species in prion diseases. Histopathology 62, 994–1001 - PubMed
-
- Alais S., Simoes S., Baas D., Lehmann S., Raposo G., Darlix J. L., Leblanc P. (2008). Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol. Cell 100, 603–618 - PubMed
-
- Apetri A. C., Surewicz K., Surewicz W. K. (2004). The effect of disease-associated mutations on the folding pathway of human prion protein. J. Biol. Chem. 279, 18008–18014 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
