

PGC-1α overexpression exacerbates β-amyloid and tau deposition in a transgenic mouse model of Alzheimer's disease
- PMID: 24398293
- PMCID: PMC3963016
- DOI: 10.1096/fj.13-236331
PGC-1α overexpression exacerbates β-amyloid and tau deposition in a transgenic mouse model of Alzheimer's disease
Abstract
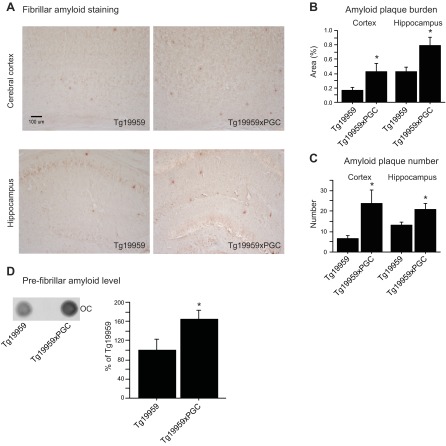
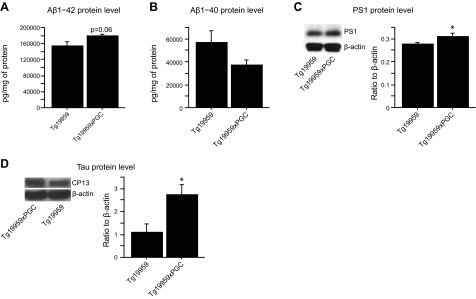
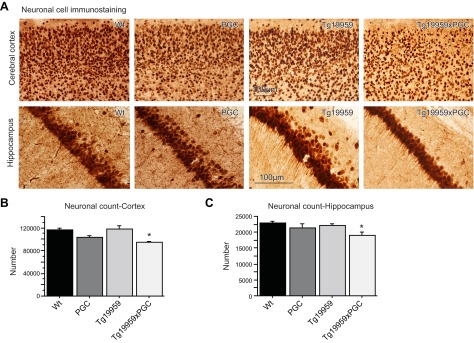
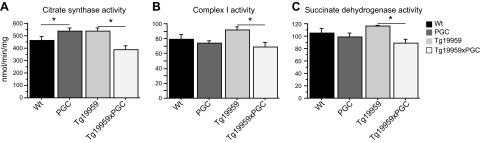
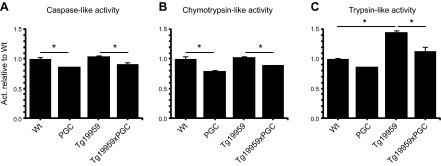
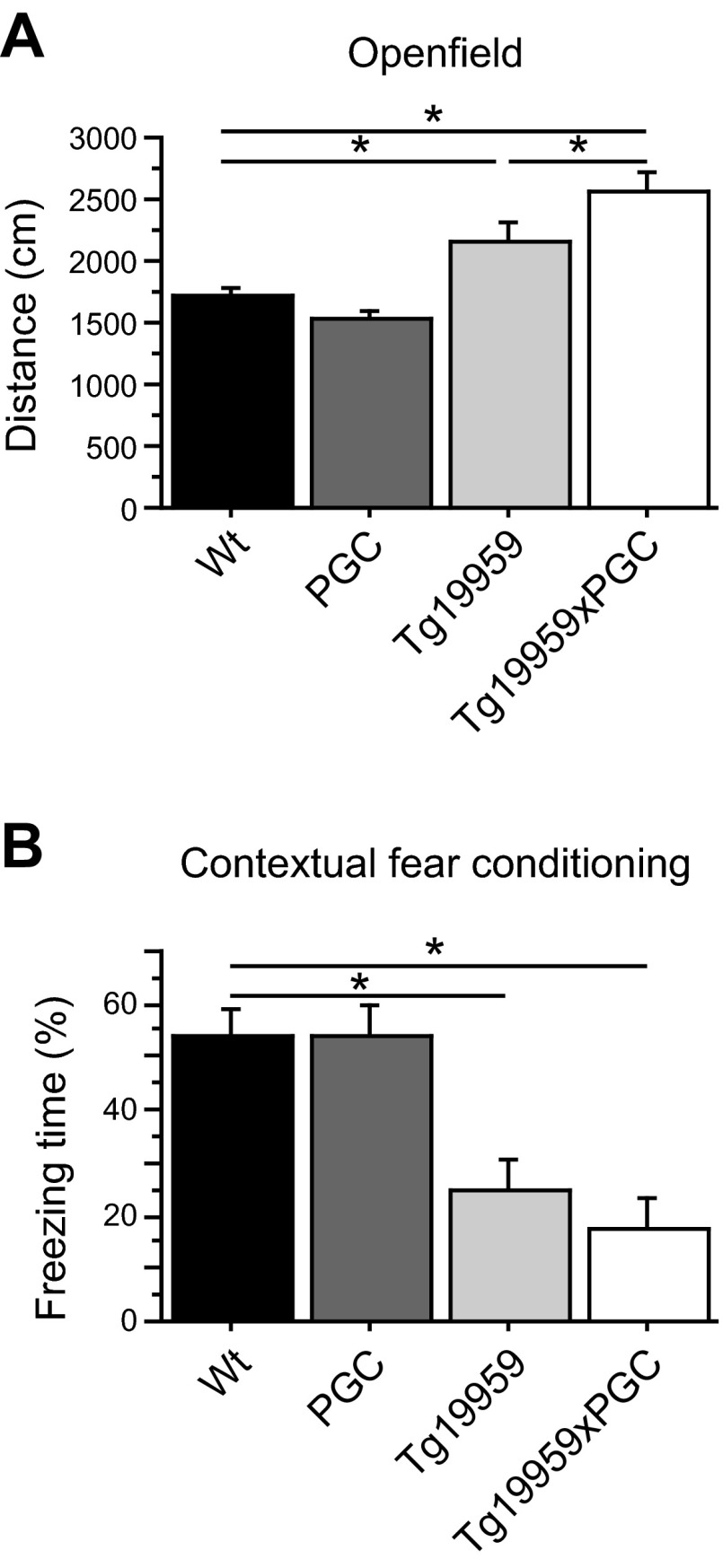
The peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) interacts with various transcription factors involved in energy metabolism and in the regulation of mitochondrial biogenesis. PGC-1α mRNA levels are reduced in a number of neurodegenerative diseases and contribute to disease pathogenesis, since increased levels ameliorate behavioral defects and neuropathology of Huntington's disease, Parkinson's disease, and amyotrophic lateral sclerosis. PGC-1α and its downstream targets are reduced both in postmortem brain tissue of patients with Alzheimer's disease (AD) and in transgenic mouse models of AD. Therefore, we investigated whether increased expression of PGC-1α would exert beneficial effects in the Tg19959 transgenic mouse model of AD; Tg19959 mice express the human amyloid precursor gene (APP) with 2 familial AD mutations and develop increased β-amyloid levels, plaque deposition, and memory deficits by 2-3 mo of age. Rather than an improvement, the cross of the Tg19959 mice with mice overexpressing human PGC-1α exacerbated amyloid and tau accumulation. This was accompanied by an impairment of proteasome activity. PGC-1α overexpression induced mitochondrial abnormalities, neuronal cell death, and an exacerbation of behavioral hyperactivity in the Tg19959 mice. These findings show that PGC-1α overexpression exacerbates the neuropathological and behavioral deficits that occur in transgenic mice with mutations in APP that are associated with human AD.
Keywords: Tg19959 mice; behavior; cell death; mitochondria.
Figures
References
-
- Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580 - PubMed
-
- Higgins G. C., Beart P. M., Shin Y. S., Chen M. J., Cheung N. S., Nagley P. (2010) Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimers. Dis. 20(Suppl. 2), S453–S473 - PubMed
-
- Lin M. T., Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 - PubMed
-
- Morais V. A., De Strooper B. (2010) Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J. Alzheimers Dis. 20(Suppl. 2), S255–S263 - PubMed
-
- Muller W. E., Eckert A., Kurz C., Eckert G. P., Leuner K. (2010) Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer's disease–therapeutic aspects. Mol. Neurobiol. 41, 159–171 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
