

Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans
- PMID: 24398540
- PMCID: PMC4676405
- DOI: 10.1007/s00467-013-2684-4
Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans
Abstract
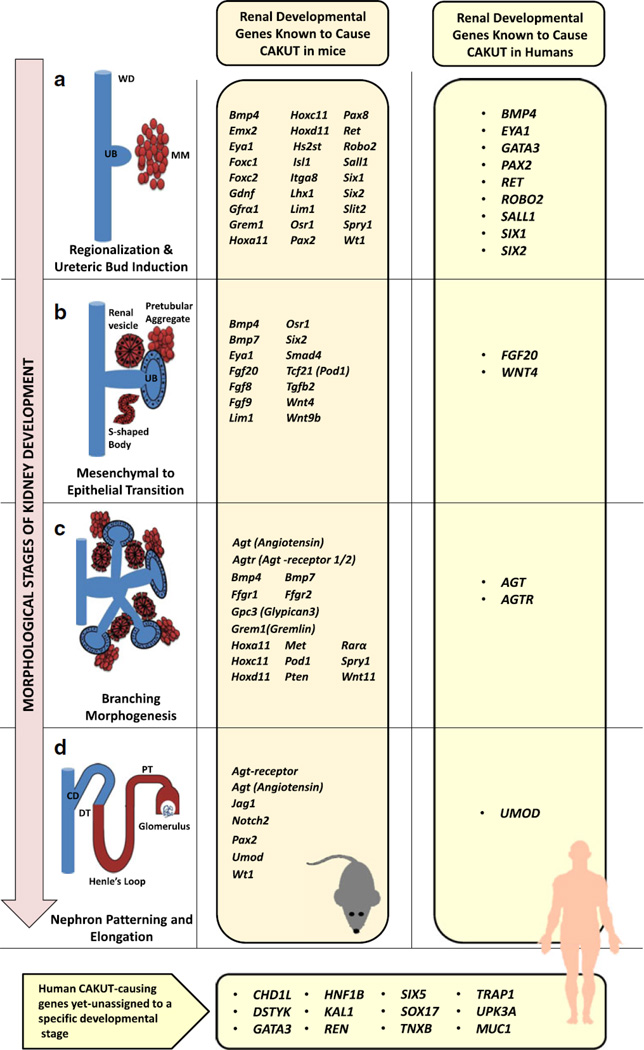
Congenital anomalies of the kidney and urinary tract (CAKUT) cover a wide range of structural malformations that result from defects in the morphogenesis of the kidney and/or urinary tract. These anomalies account for about 40-50 % of children with chronic kidney disease worldwide. Knowledge from genetically modified mouse models suggests that single gene mutations in renal developmental genes may lead to CAKUT in humans. However, until recently, only a handful of CAKUT-causing genes were reported, most of them in familial syndromic cases. Recent findings suggest that CAKUT may arise from mutations in a multitude of different single gene causes. We focus here on single-gene causes of CAKUT and their developmental origin. Currently, more than 20 monogenic CAKUT-causing genes have been identified. High-throughput sequencing techniques make it likely that additional CAKUT-causing genes will be identified in the near future.
Figures
References
-
- Schedl A. Renal abnormalities and their developmental origin. Nature Reviews Genetics. 2007;8:791–802. - PubMed
-
- (2008) North American Pediatric Renal Transplant Cooperative Study (NAPRTCS) 2008 Annual report. Rockville, MD: The EMMES Corporation; 2008.
-
- Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. 1995;9:358–364. - PubMed
-
- Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Human molecular genetics. 1999;8:2001–2008. - PubMed
-
- Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:157–164. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
