

Advances in motor neurone disease
- PMID: 24399773
- PMCID: PMC3883149
- DOI: 10.1177/0141076813511451
Advances in motor neurone disease
Abstract
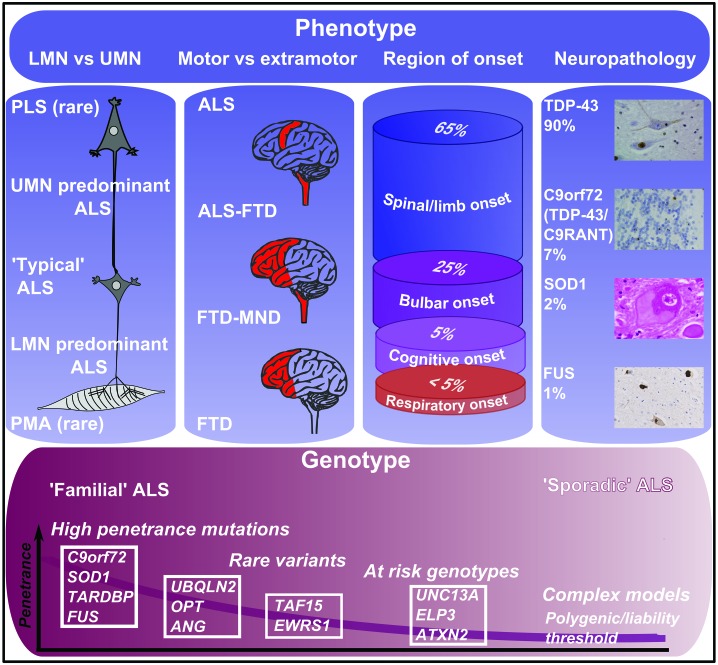
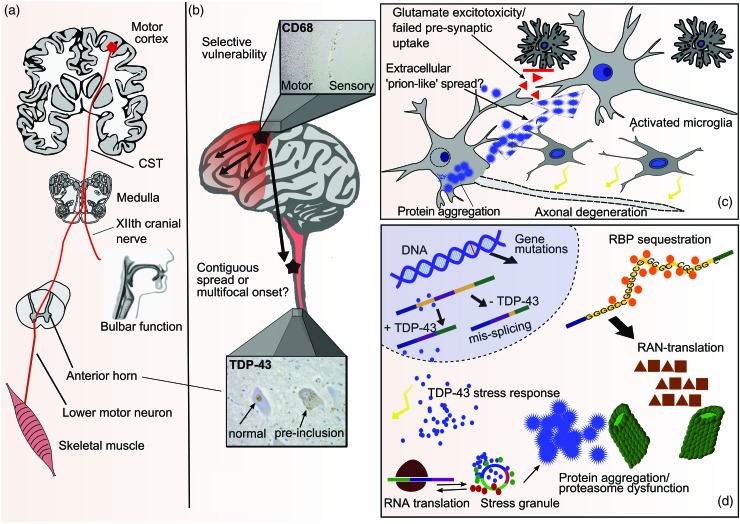
Motor neurone disease (MND), the commonest clinical presentation of which is amyotrophic lateral sclerosis (ALS), is regarded as the most devastating of adult-onset neurodegenerative disorders. The last decade has seen major improvements in patient care, but also rapid scientific advances, so that rational therapies based on key pathogenic mechanisms now seem plausible. ALS is strikingly heterogeneous in both its presentation, with an average one-year delay from first symptoms to diagnosis, and subsequent rate of clinical progression. Although half of patients succumb within 3-4 years of symptom onset, typically through respiratory failure, a significant minority survives into a second decade. Although an apparently sporadic disorder for most patients, without clear environmental triggers, recent genetic studies have identified disease-causing mutations in genes in several seemingly disparate functional pathways, so that motor neuron degeneration may need to be understood as a common final pathway with a number of upstream causes. This apparent aetiological and clinical heterogeneity suggests that therapeutic studies should include detailed biomarker profiling, and consider genetic as well as clinical stratification. The most common mutation, accounting for 10% of all Western hemisphere ALS, is a hexanucleotide repeat expansion in C9orf72. This and several other genes implicate altered RNA processing and protein degradation pathways in the core of ALS pathogenesis. A major gap remains in understanding how such fundamental processes appear to function without obvious deficit in the decades prior to symptom emergence, and the study of pre-symptomatic gene carriers is an important new initiative.
Keywords: RNA; TDP-43; amyotrophic lateral sclerosis; anterior horn cell; autophagy; frontotemporal dementia; neurodegeneration; protein aggregation.
Figures
References
-
- Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011; 377: 942–55 - PubMed
-
- Phukan J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2012; 83: 102–8 - PubMed
-
- Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 293–9 - PubMed
-
- de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 2008; 119: 497–503 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
