

Evidence for eIF2α phosphorylation-independent effects of GSK2656157, a novel catalytic inhibitor of PERK with clinical implications
- PMID: 24401334
- PMCID: PMC3979916
- DOI: 10.4161/cc.27726
Evidence for eIF2α phosphorylation-independent effects of GSK2656157, a novel catalytic inhibitor of PERK with clinical implications
Abstract
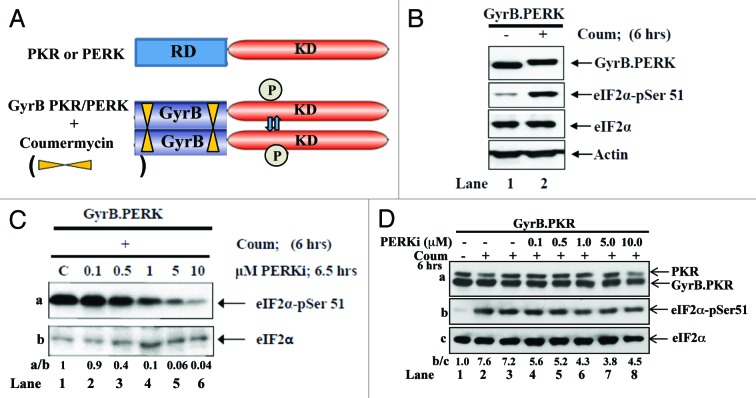
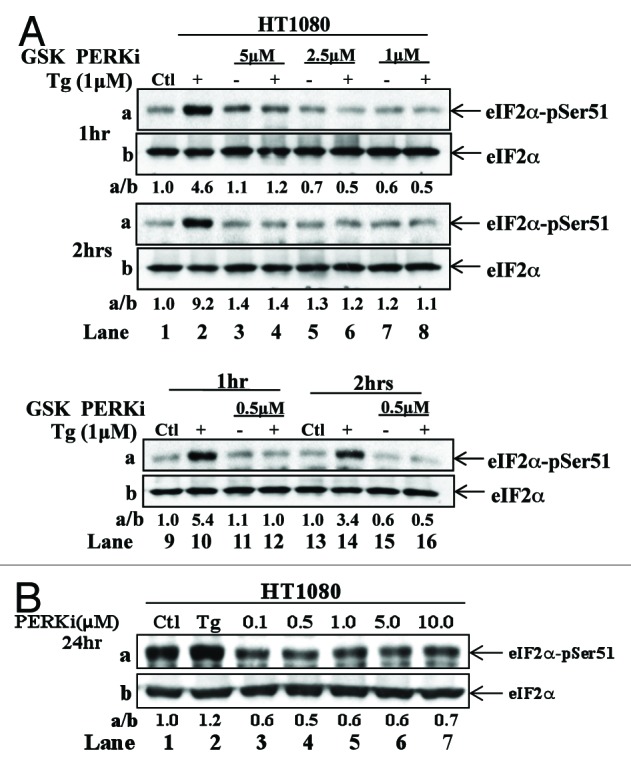
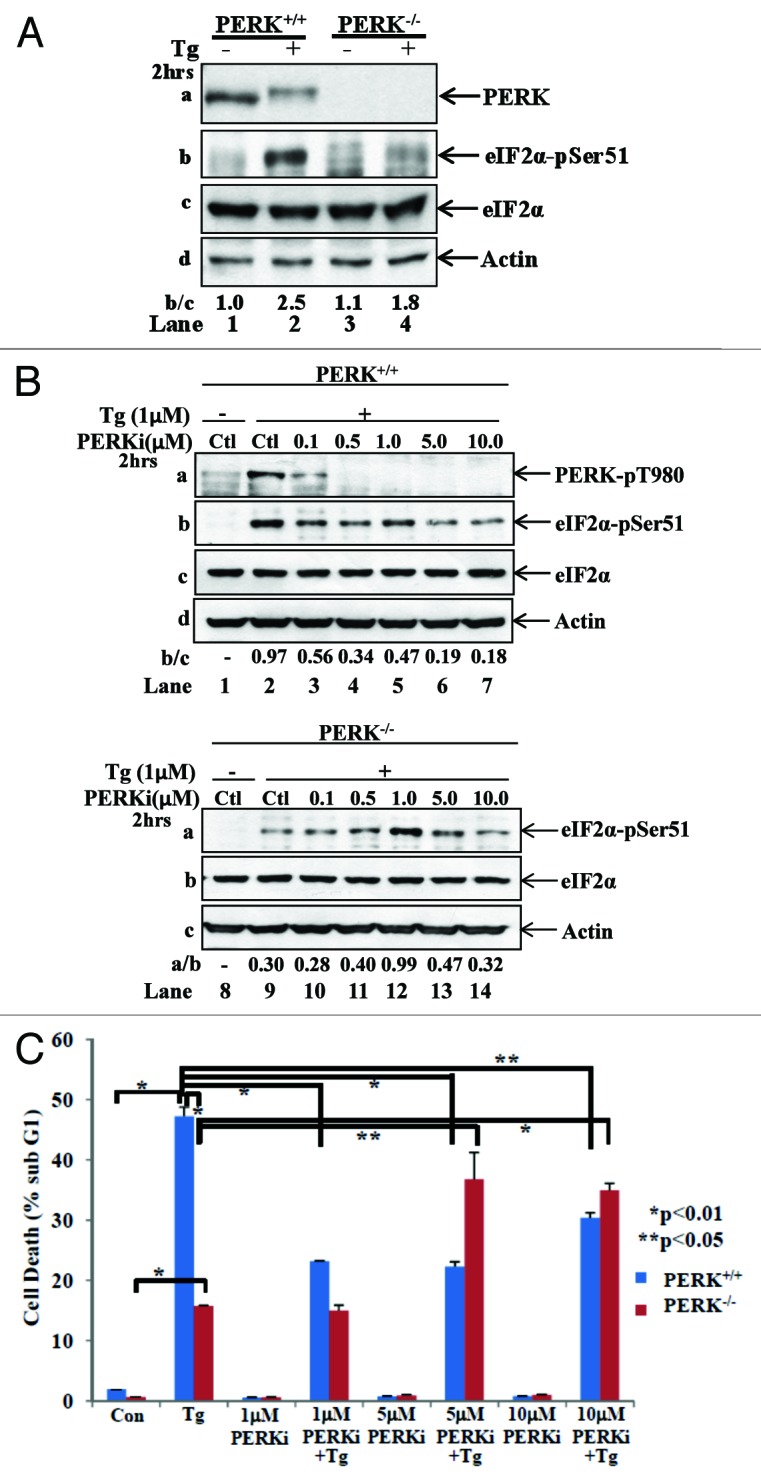
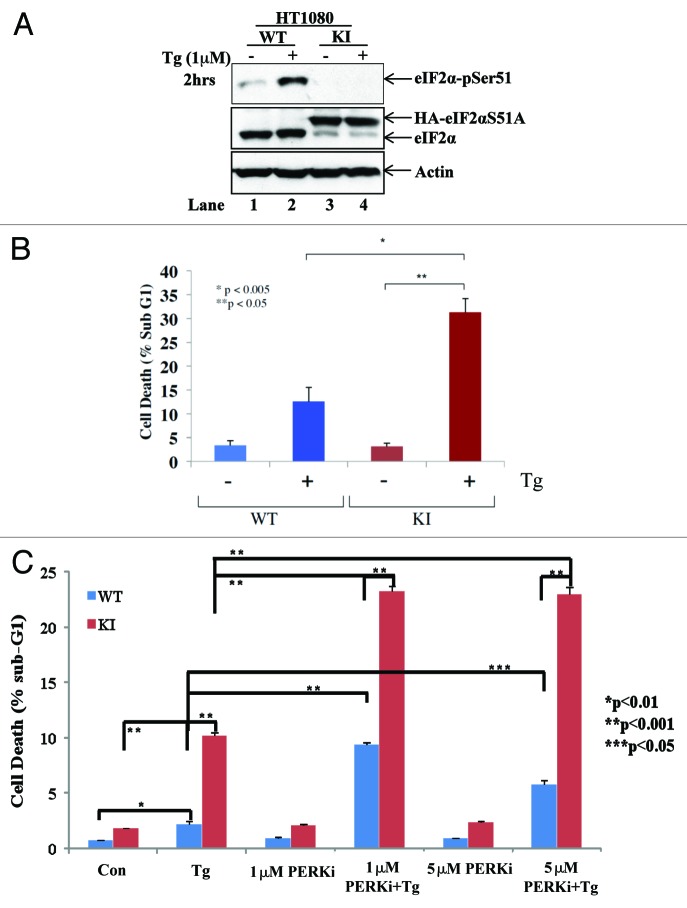
The endoplasmic reticulum (ER)-resident protein kinase PERK is a major component of the unfolded protein response (UPR), which promotes the adaptation of cells to various forms of stress. PERK phosphorylates the α subunit of the translation initiation factor eIF2 at serine 51, a modification that plays a key role in the regulation of mRNA translation in stressed cells. Several studies have demonstrated that the PERK-eIF2α phosphorylation pathway maintains insulin biosynthesis and glucose homeostasis, facilitates tumor formation and decreases the efficacy of tumor treatment with chemotherapeutic drugs. Recently, a selective catalytic PERK inhibitor termed GSK2656157 has been developed with anti-tumor properties in mice. Herein, we provide evidence that inhibition of PERK activity by GSK2656157 does not always correlate with inhibition of eIF2α phosphorylation. Also, GSK2656157 does not always mimic the biological effects of the genetic inactivation of PERK. Furthermore, cells treated with GSK2656157 increase eIF2α phosphorylation as a means to compensate for the loss of PERK. Using human tumor cells impaired in eIF2α phosphorylation, we demonstrate that GSK2656157 induces ER stress-mediated death suggesting that the drug acts independent of the inhibition of eIF2α phosphorylation. We conclude that GSK2656157 might be a useful compound to dissect pathways that compensate for the loss of PERK and/or identify PERK pathways that are independent of eIF2α phosphorylation.
Keywords: PERK/PEK kinase; endoplasmic reticulum stress; mRNA translation; pharmacological inhibitors; protein phosphorylation; translation initiation factor eIF2; tumorigenesis; unfolded protein response.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources