

Implications of prion adaptation and evolution paradigm for human neurodegenerative diseases
- PMID: 24401672
- PMCID: PMC7030914
- DOI: 10.4161/pri.27661
Implications of prion adaptation and evolution paradigm for human neurodegenerative diseases
Abstract
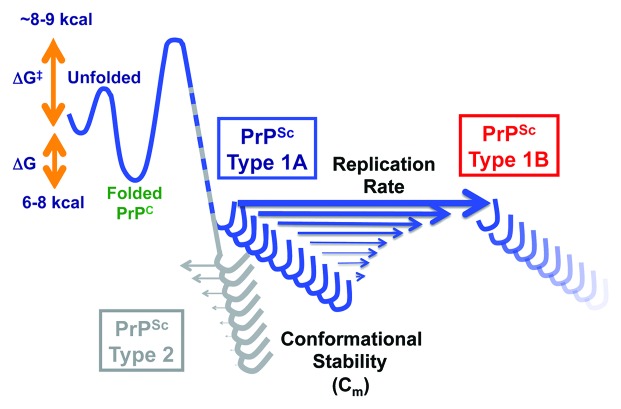
There is a growing body of evidence indicating that number of human neurodegenerative diseases, including Alzheimer disease, Parkinson disease, fronto-temporal dementias, and amyotrophic lateral sclerosis, propagate in the brain via prion-like intercellular induction of protein misfolding. Prions cause lethal neurodegenerative diseases in humans, the most prevalent being sporadic Creutzfeldt-Jakob disease (sCJD); they self-replicate and spread by converting the cellular form of prion protein (PrP(C)) to a misfolded pathogenic conformer (PrP(Sc)). The extensive phenotypic heterogeneity of human prion diseases is determined by polymorphisms in the prion protein gene, and by prion strain-specific conformation of PrP(Sc). Remarkably, even though informative nucleic acid is absent, prions may undergo rapid adaptation and evolution in cloned cells and upon crossing the species barrier. In the course of our investigation of this process, we isolated distinct populations of PrP(Sc) particles that frequently co-exist in sCJD. The human prion particles replicate independently and undergo competitive selection of those with lower initial conformational stability. Exposed to mutant substrate, the winning PrP(Sc) conformers are subject to further evolution by natural selection of the subpopulation with the highest replication rate due to the lowest stability. Thus, the evolution and adaptation of human prions is enabled by a dynamic collection of distinct populations of particles, whose evolution is governed by the selection of progressively less stable, faster replicating PrP(Sc) conformers. This fundamental biological mechanism may explain the drug resistance that some prions gained after exposure to compounds targeting PrP(Sc). Whether the phenotypic heterogeneity of other neurodegenerative diseases caused by protein misfolding is determined by the spectrum of misfolded conformers (strains) remains to be established. However, the prospect that these conformers may evolve and adapt by a prion-like mechanism calls for the reevaluation of therapeutic strategies that target aggregates of misfolded proteins, and argues for new therapeutic approaches that will focus on prior pathogenetic steps.
Figures
References
-
- Gibbs CJ Jr., Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, Matthews WB. . Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 1968; 161:388 - 9; http://dx.doi.org/ 10.1126/science.161.3839.388; PMID: 5661299 - DOI - PubMed
-
- Brown P, Gibbs CJ Jr., Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. . Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994; 35:513 - 29; http://dx.doi.org/ 10.1002/ana.410350504; PMID: 8179297 - DOI - PubMed
-
- Prusiner SB. . Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/ 10.1126/science.6801762; PMID: 6801762 - DOI - PubMed
-
- Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. . Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/ 10.1073/pnas.90.23.10962; PMID: 7902575 - DOI - PMC - PubMed
-
- Prusiner SB. . Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/ 10.1073/pnas.95.23.13363; PMID: 9811807 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials