

Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992-2012
- PMID: 24402832
- PMCID: PMC3930393
- DOI: 10.1002/cam4.161
Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992-2012
Abstract
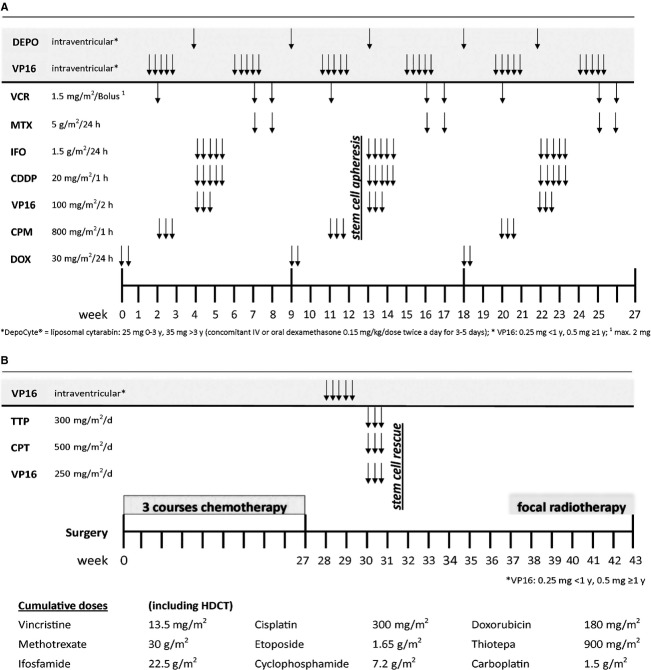
Atypical teratoid rhabdoid tumors (ATRTs) are recently defined highly aggressive embryonal central nervous system tumors with a poor prognosis and no definitive guidelines for treatment. We report on the importance of an initial correct diagnosis and disease-specific therapy on outcome in 22 consecutive patients and propose a new treatment strategy. From 1992 to 2012, nine patients initially diagnosed correctly as ATRT (cohort A, median age 24 months) were treated according to an intensive multimodal regimen (MUV-ATRT) consisting of three 9-week courses of a dose-dense regimen including doxorubicin, cyclophosphamide, vincristine, ifosfamide, cisplatin, etoposide, and methotrexate augmented with intrathecal therapy, followed by high-dose chemotherapy (HDCT) and completed with local radiotherapy. Thirteen patients were treated differently (cohort B, median age 30 months) most of whom according to protocols in use for their respective diagnoses. As of July 2013, 5-year overall survival (OS) and event-free survival (EFS) for all 22 consecutive patients was 56.3 ± 11.3% and 52.9 ± 11.0%, respectively. For MUV-ATRT regimen-treated patients (cohort A) 5-year OS was 100% and EFS was 88.9 ± 10.5%. For patients treated differently (cohort B) 5-year OS and EFS were 28.8 ± 13.1%. All nine MUV-ATRT regimen-treated patients are alive for a median of 76 months (range: 16-197), eight in first complete remission. Our results compare favorably to previously published data. The drug combination and sequence used in the proposed MUV-ATRT regimen appear to be efficacious in preventing early relapses also in young children with M1-M3 stage disease allowing postponement of radiotherapy until after HDCT.
Keywords: ATRT; delayed local radiotherapy; high-dose chemotherapy; improved survival; multimodal therapy.
© 2013 The Authors. Cancer Medicine published by John Wiley & Sons Ltd.
Figures

References
-
- Woehrer A, Slavc I, Waldhoer T, Heinzl H, Zielonke N, Czech T, et al. Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996–2006. Cancer. 2010;116:5725–5732. - PubMed
-
- Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer. 1978;41:1937–1948. - PubMed
-
- Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J. Neurosurg. 1996;85:56–65. - PubMed
-
- Kleihues P, Cavanee WK. Pathology and genetics of tumours of the nervous system. 3rd ed. Lyon, France: IARC Press; 2000.
-
- Bourdeaut F, Lequin D, Brugieres L, Reynaud S, Dufour C, Doz F, et al. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin. Cancer Res. 2011;17:31–38. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
