

Synthetic lethality screen identifies RPS6KA2 as modifier of epidermal growth factor receptor activity in pancreatic cancer
- PMID: 24403857
- PMCID: PMC3884526
- DOI: 10.1593/neo.131660
Synthetic lethality screen identifies RPS6KA2 as modifier of epidermal growth factor receptor activity in pancreatic cancer
Abstract
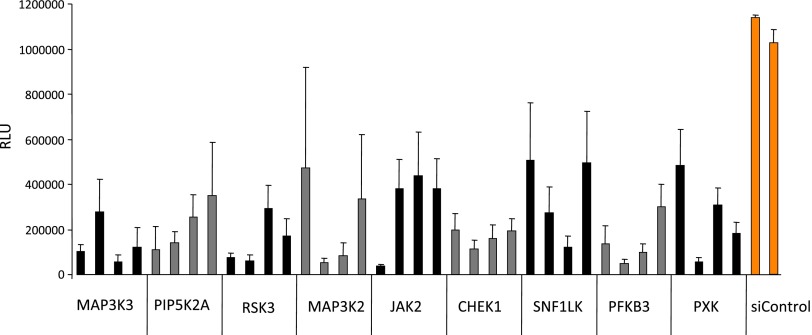
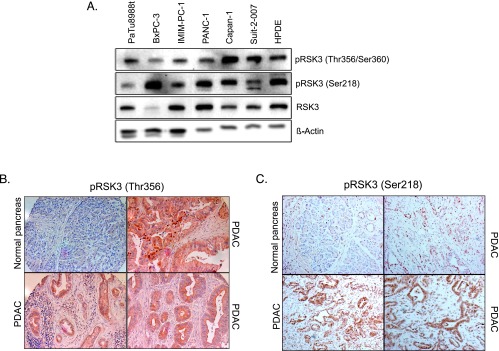
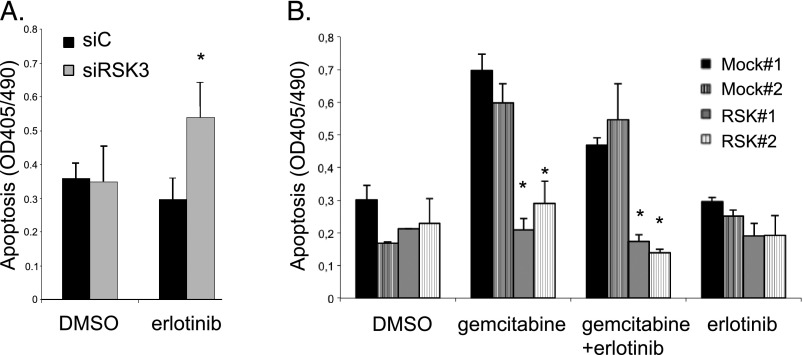
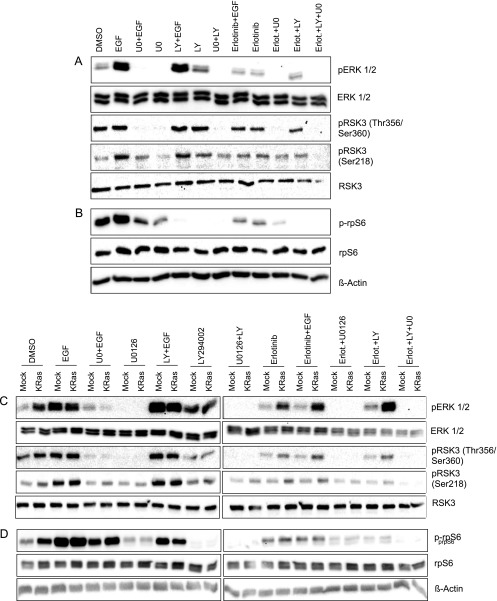
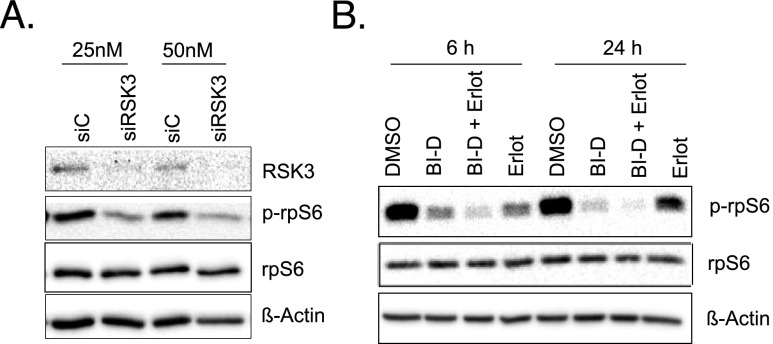
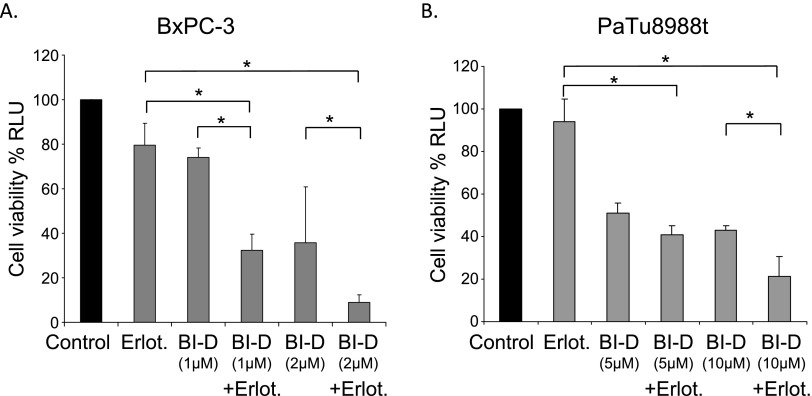
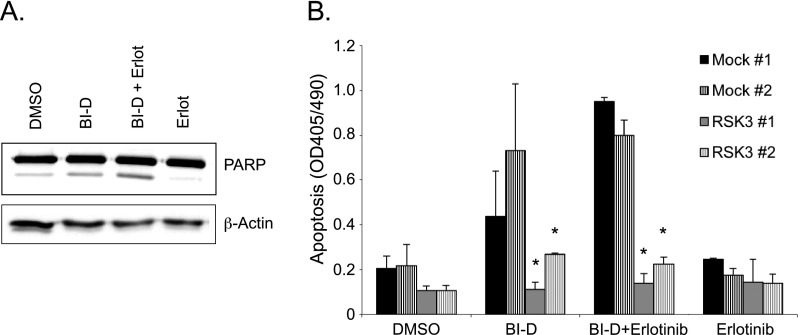
Pancreatic cancer is characterized by a high degree of resistance to chemotherapy. Epidermal growth factor receptor (EGFR) inhibition using the small-molecule inhibitor erlotinib was shown to provide a small survival benefit in a subgroup of patients. To identify kinases whose inhibition acts synergistically with erlotinib, we employed a kinome-wide small-interfering RNA (siRNA)-based loss-of-function screen in the presence of erlotinib. Of 779 tested kinases, we identified several targets whose inhibition acted synergistically lethal with EGFR inhibition by erlotinib, among them the S6 kinase ribosomal protein S6 kinase 2 (RPS6KA2)/ribosomal S6 kinase 3. Activated RPS6KA2 was expressed in approximately 40% of 123 human pancreatic cancer tissues. RPS6KA2 was shown to act downstream of EGFR/RAS/mitogen-activated protein kinase kinase (MEK)/extracellular-signal regulated kinase (ERK) signaling and was activated by EGF independently of the presence of KRAS mutations. Knockdown of RPS6KA2 by siRNA led to increased apoptosis only in the presence of erlotinib, whereas RPS6KA2 activation or overexpression rescued from erlotinib- and gemcitabine-induced apoptosis. This effect was at least in part mediated by downstream activation of ribosomal protein S6. Genetic as well as pharmacological inhibition of RPS6KA2 by the inhibitor BI-D1870 acted synergistically with erlotinib. By applying this synergistic lethality screen using a kinome-wide RNA interference-library approach, we identified RPS6KA2 as potential drug target whose inhibition synergistically enhanced the effect of erlotinib on tumor cell survival. This kinase therefore represents a promising drug candidate suitable for the development of novel inhibitors for pancreatic cancer therapy.
Figures
Similar articles
-
Insights into erlotinib action in pancreatic cancer cells using a combined experimental and mathematical approach.World J Gastroenterol. 2012 Nov 21;18(43):6226-34. doi: 10.3748/wjg.v18.i43.6226. World J Gastroenterol. 2012. PMID: 23180942 Free PMC article.
-
Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors.Mol Cancer Ther. 2006 Nov;5(11):2676-84. doi: 10.1158/1535-7163.MCT-06-0166. Mol Cancer Ther. 2006. PMID: 17121914
-
Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells.Clin Cancer Res. 2011 May 1;17(9):2744-56. doi: 10.1158/1078-0432.CCR-10-2214. Epub 2011 Mar 8. Clin Cancer Res. 2011. PMID: 21385921 Free PMC article.
-
Targeting EGFR in pancreatic cancer treatment.Curr Drug Targets. 2012 Jun;13(6):802-10. doi: 10.2174/138945012800564158. Curr Drug Targets. 2012. PMID: 22458527 Review.
-
Erlotinib in cancer treatment.Ann Oncol. 2007 Jun;18 Suppl 6:vi35-41. doi: 10.1093/annonc/mdm222. Ann Oncol. 2007. PMID: 17591829 Review.
Cited by
-
BI-D1870 Induces Mitotic Dysfunction and Apoptosis in Neuroblastoma by Regulating the PI3K-Akt-mTORC1 Signal Axis.Cancers (Basel). 2023 Mar 28;15(7):2023. doi: 10.3390/cancers15072023. Cancers (Basel). 2023. PMID: 37046682 Free PMC article.
-
Exploration of an Integrative Prognostic Model of Radiogenomics Features With Underlying Gene Expression Patterns in Clear Cell Renal Cell Carcinoma.Front Oncol. 2021 Mar 8;11:640881. doi: 10.3389/fonc.2021.640881. eCollection 2021. Front Oncol. 2021. PMID: 33763374 Free PMC article.
-
MptpB Promotes Mycobacteria Survival by Inhibiting the Expression of Inflammatory Mediators and Cell Apoptosis in Macrophages.Front Cell Infect Microbiol. 2018 May 25;8:171. doi: 10.3389/fcimb.2018.00171. eCollection 2018. Front Cell Infect Microbiol. 2018. PMID: 29888212 Free PMC article.
-
Genome-wide association study of café-au-lait macule number in neurofibromatosis type 1.Mol Genet Genomic Med. 2020 Oct;8(10):e1400. doi: 10.1002/mgg3.1400. Epub 2020 Aug 31. Mol Genet Genomic Med. 2020. PMID: 32869517 Free PMC article.
-
Identification of a transcriptomic signature with excellent survival prediction for squamous cell carcinoma of the cervix.Am J Cancer Res. 2020 May 1;10(5):1534-1547. eCollection 2020. Am J Cancer Res. 2020. PMID: 32509396 Free PMC article.
References
-
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. - PubMed
-
- Troiani T, Martinelli E, Capasso A, Morgillo F, Orditura M, De Vita F, Ciardiello F. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets. 2012;13(6):802–810. - PubMed
-
- Michl P, Gress TM. Current concepts and novel targets in advanced pancreatic cancer. Gut. 2013;62:317–326. - PubMed
-
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. - PubMed
-
- Heinemann V, Haas M, Boeck S. Systemic treatment of advanced pancreatic cancer. Cancer Treat Rev. 2012;38(7):843–853. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous