

The impact of human hyperekplexia mutations on glycine receptor structure and function
- PMID: 24405574
- PMCID: PMC3895786
- DOI: 10.1186/1756-6606-7-2
The impact of human hyperekplexia mutations on glycine receptor structure and function
Abstract
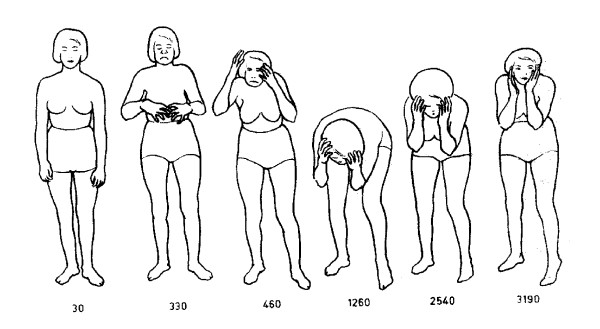
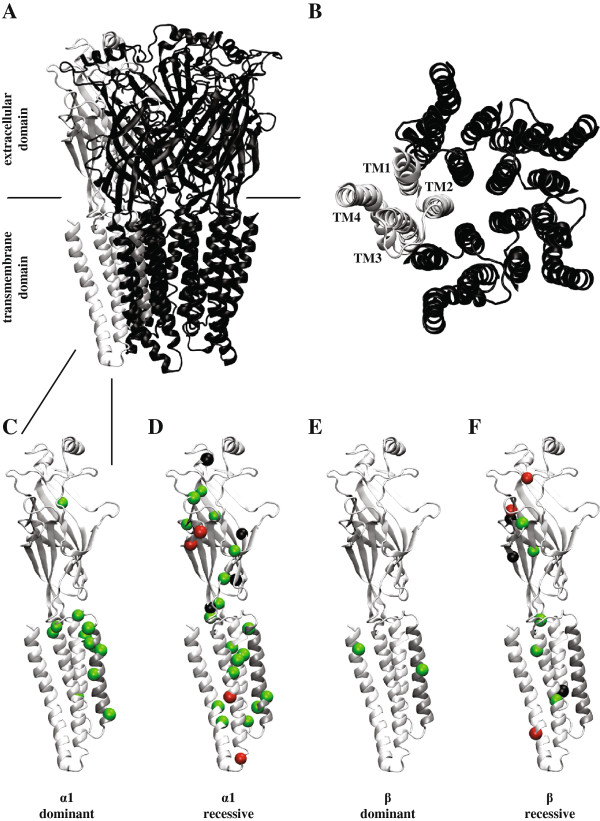
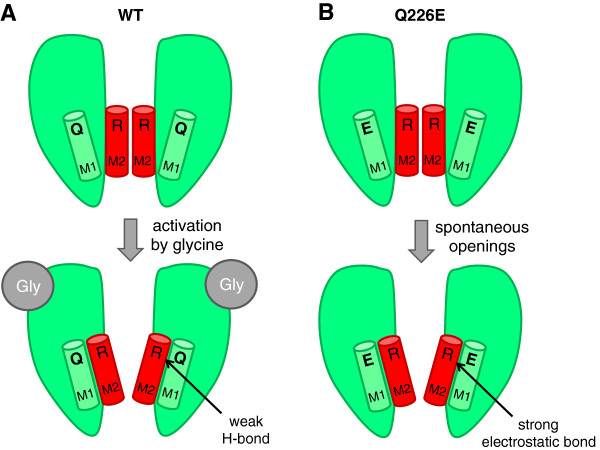
Hyperekplexia is a rare neurological disorder characterized by neonatal hypertonia, exaggerated startle responses to unexpected stimuli and a variable incidence of apnoea, intellectual disability and delays in speech acquisition. The majority of motor defects are successfully treated by clonazepam. Hyperekplexia is caused by hereditary mutations that disrupt the functioning of inhibitory glycinergic synapses in neuromotor pathways of the spinal cord and brainstem. The human glycine receptor α1 and β subunits, which predominate at these synapses, are the major targets of mutations. International genetic screening programs, that together have analysed several hundred probands, have recently generated a clear picture of genotype-phenotype correlations and the prevalence of different categories of hyperekplexia mutations. Focusing largely on this new information, this review seeks to summarise the effects of mutations on glycine receptor structure and function and how these functional alterations lead to hyperekplexia.
Figures
References
-
- Suhren O, Bruyn GW, Tuynman JA. Hyperexplexia - a hereditary startle syndrome. J Neurol Sci. 1966;3:577–605. doi: 10.1016/0022-510X(66)90047-5. - DOI
-
- Thomas RH, Harvey RJ, Rees MI. Hyperekplexia: stiffness, startle and syncope. J Pediatr Neurol. 2010;8:11–14.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
