

Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson's model
- PMID: 24407237
- PMCID: PMC4040705
- DOI: 10.1038/cddis.2013.513
Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson's model
Abstract
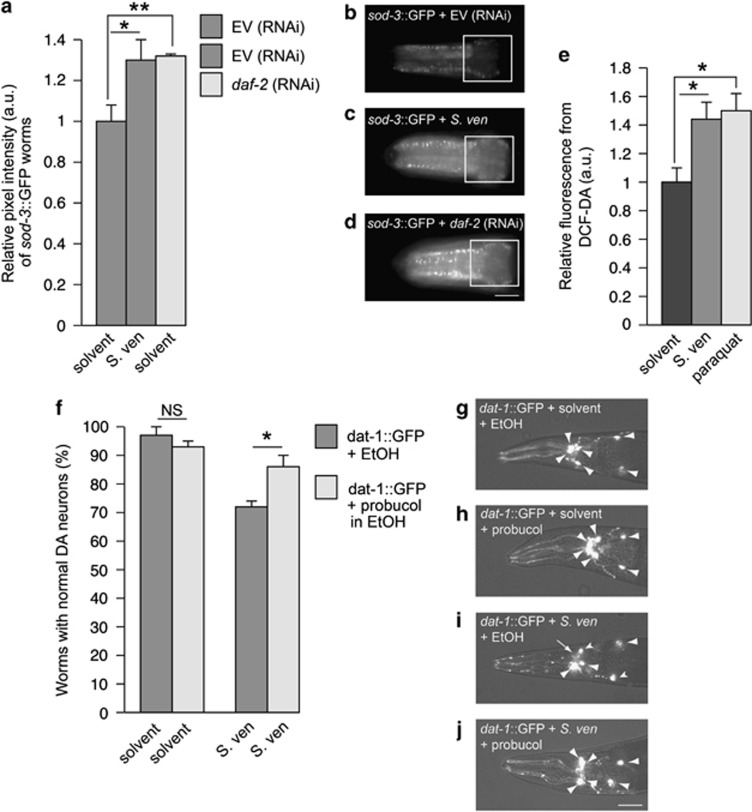
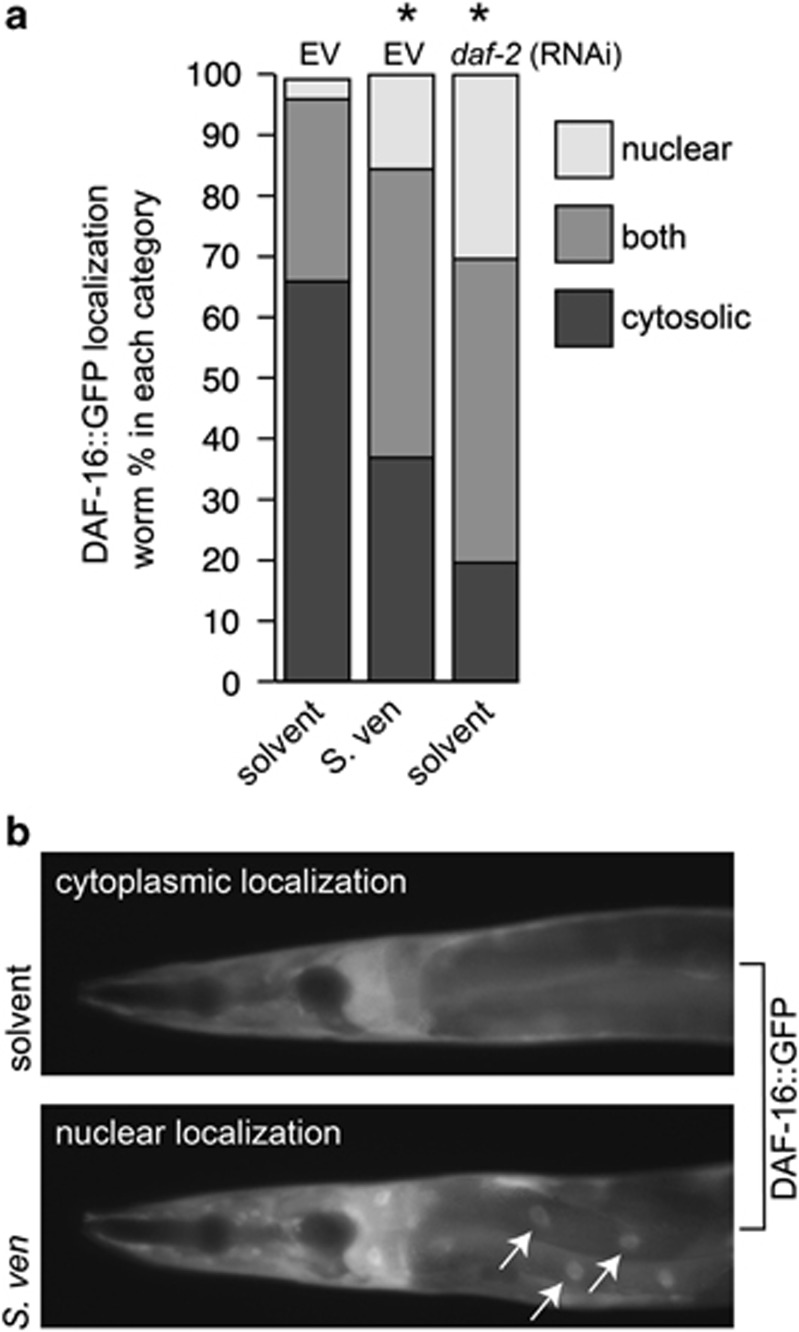
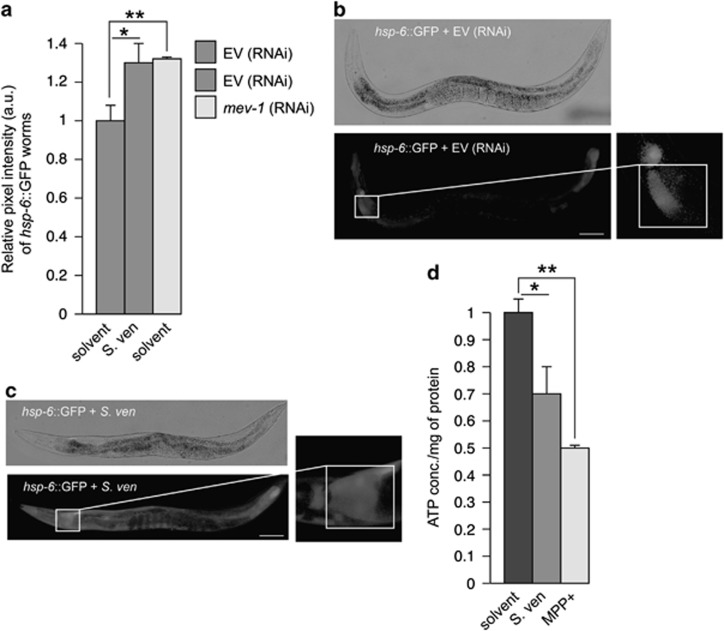
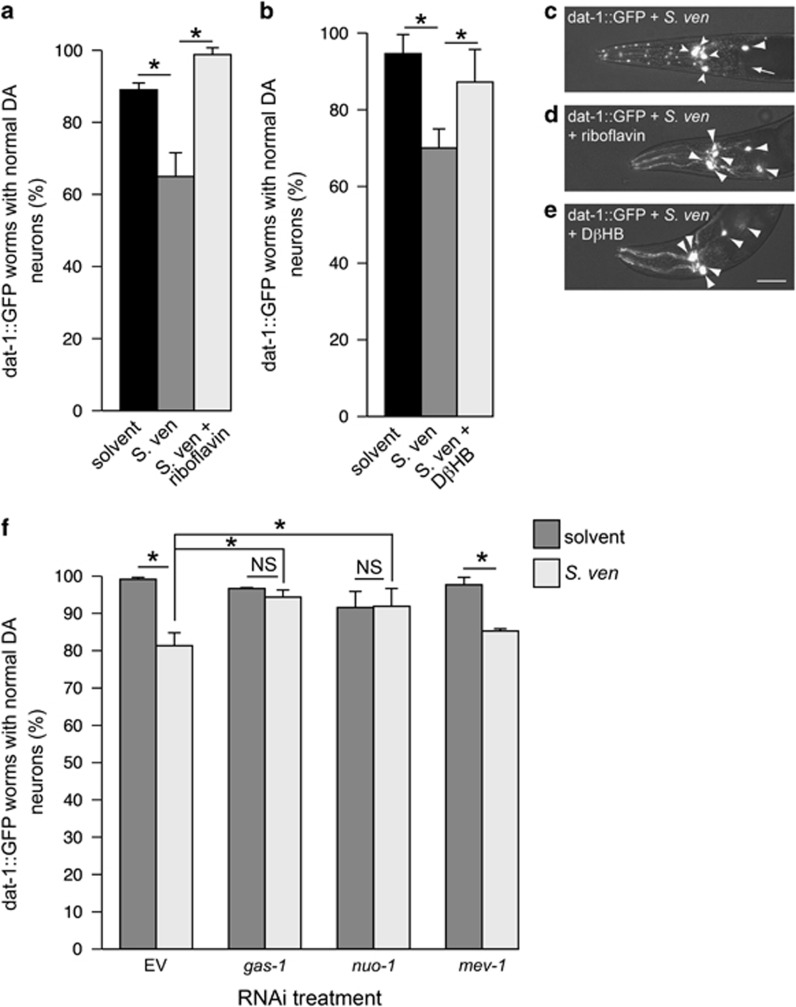
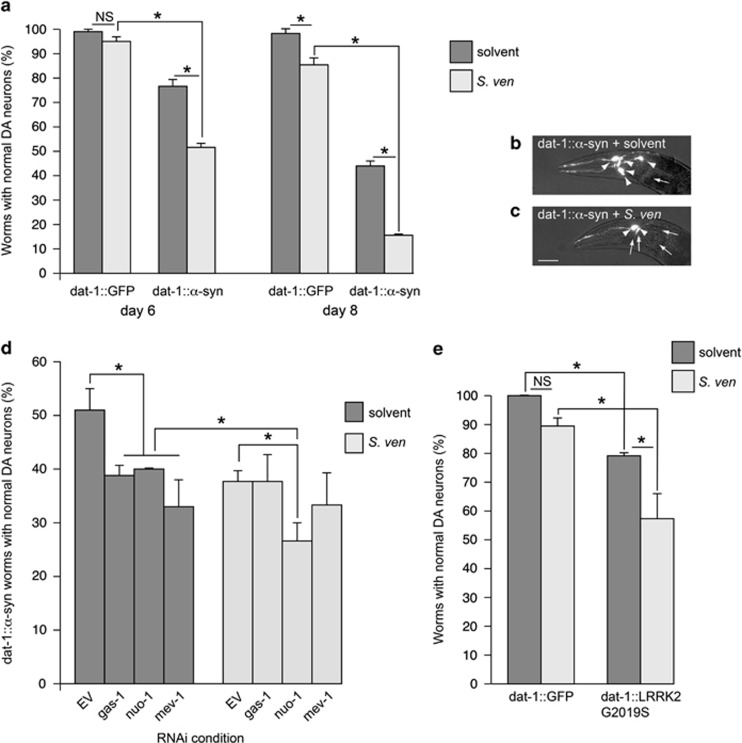
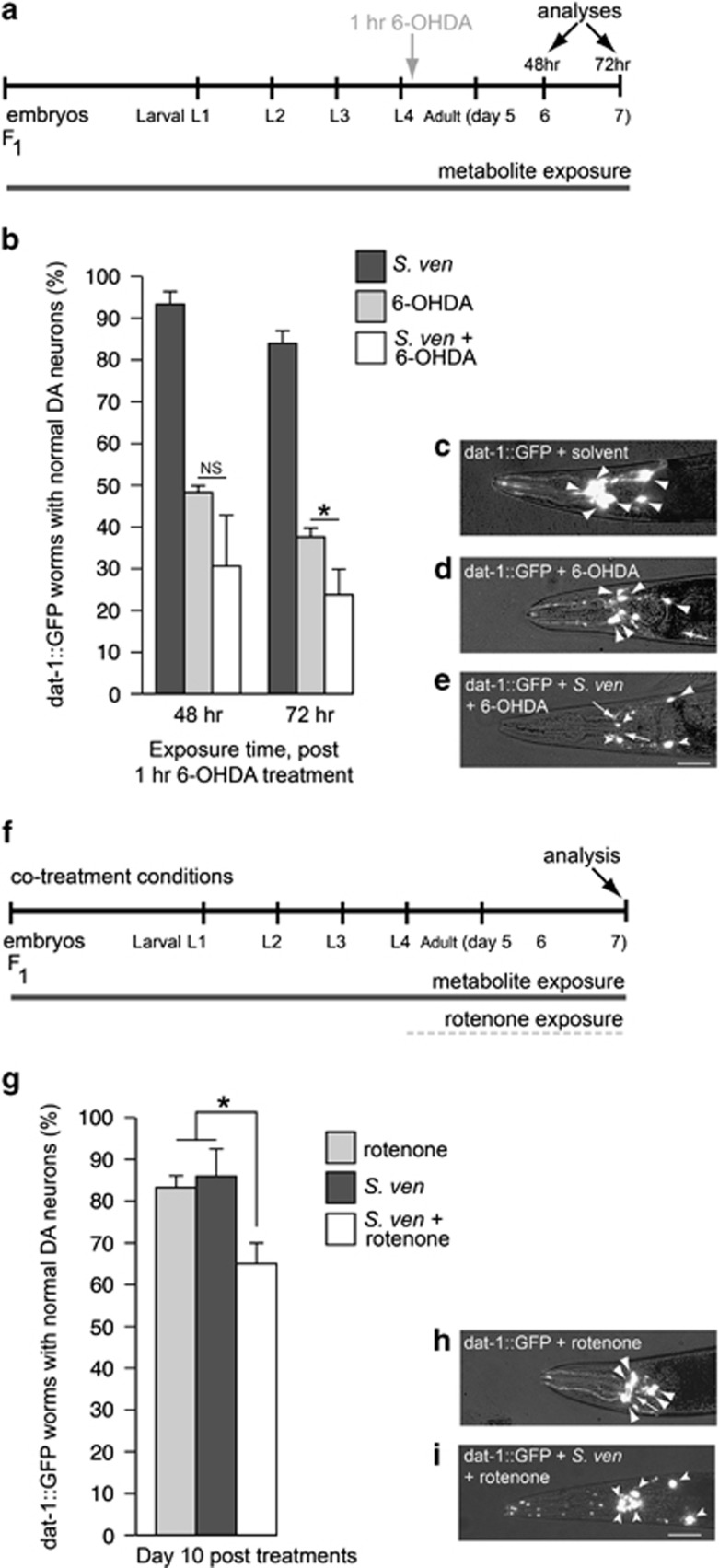
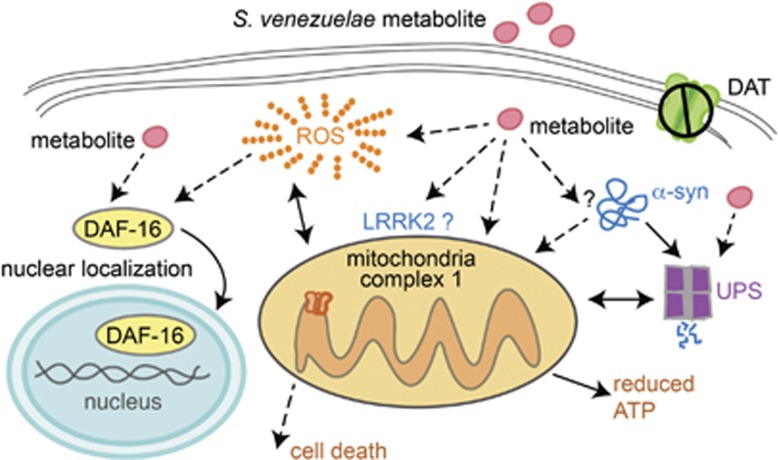
Genetic and idiopathic forms of Parkinson's disease (PD) are characterized by loss of dopamine (DA) neurons and typically the formation of protein inclusions containing the alpha-synuclein (α-syn) protein. Environmental contributors to PD remain largely unresolved but toxins, such as paraquat or rotenone, represent well-studied enhancers of susceptibility. Previously, we reported that a bacterial metabolite produced by Streptomyces venezuelae caused age- and dose-dependent DA neurodegeneration in Caenorhabditis elegans and human SH-SY5Y neurons. We hypothesized that this metabolite from a common soil bacterium could enhance neurodegeneration in combination with PD susceptibility gene mutations or toxicants. Here, we report that exposure to the metabolite in C. elegans DA neurons expressing human α-syn or LRRK2 G2019S exacerbates neurodegeneration. Using the PD toxin models 6-hydroxydopamine and rotenone, we demonstrate that exposure to more than one environmental risk factor has an additive effect in eliciting DA neurodegeneration. Evidence suggests that PD-related toxicants cause mitochondrial dysfunction, thus we examined the impact of the metabolite on mitochondrial activity and oxidative stress. An ex vivo assay of C. elegans extracts revealed that this metabolite causes excessive production of reactive oxygen species. Likewise, enhanced expression of a superoxide dismutase reporter was observed in vivo. The anti-oxidant probucol fully rescued metabolite-induced DA neurodegeneration, as well. Interestingly, the stress-responsive FOXO transcription factor DAF-16 was activated following exposure to the metabolite. Through further mechanistic analysis, we discerned the mitochondrial defects associated with metabolite exposure included adenosine triphosphate impairment and upregulation of the mitochondrial unfolded protein response. Metabolite-induced toxicity in DA neurons was rescued by complex I activators. RNA interference (RNAi) knockdown of mitochondrial complex I subunits resulted in rescue of metabolite-induced toxicity in DA neurons. Taken together, our characterization of cellular responses to the S. venezuelae metabolite indicates that this putative environmental trigger of neurotoxicity may cause cell death, in part, through mitochondrial dysfunction and oxidative stress.
Figures
Similar articles
-
Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson's models.Cell Death Dis. 2018 May 1;9(5):555. doi: 10.1038/s41419-018-0619-5. Cell Death Dis. 2018. PMID: 29748634 Free PMC article.
-
Application of a C. elegans dopamine neuron degeneration assay for the validation of potential Parkinson's disease genes.J Vis Exp. 2008 Jul 18;(17):835. doi: 10.3791/835. J Vis Exp. 2008. PMID: 19066512 Free PMC article.
-
Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson's Disease.J Neurosci. 2017 Nov 15;37(46):11085-11100. doi: 10.1523/JNEUROSCI.1294-17.2017. Epub 2017 Oct 13. J Neurosci. 2017. PMID: 29030433 Free PMC article.
-
Reprint of: revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease-resemblance to the effect of amphetamine drugs of abuse.Free Radic Biol Med. 2013 Sep;62:186-201. doi: 10.1016/j.freeradbiomed.2013.05.042. Epub 2013 Jun 3. Free Radic Biol Med. 2013. PMID: 23743292 Review.
-
No Country for Old Worms: A Systematic Review of the Application of C. elegans to Investigate a Bacterial Source of Environmental Neurotoxicity in Parkinson's Disease.Metabolites. 2018 Oct 29;8(4):70. doi: 10.3390/metabo8040070. Metabolites. 2018. PMID: 30380609 Free PMC article. Review.
Cited by
-
LRRK2 mutations and neurotoxicant susceptibility.Exp Biol Med (Maywood). 2015 Jun;240(6):752-9. doi: 10.1177/1535370215579162. Epub 2015 Apr 16. Exp Biol Med (Maywood). 2015. PMID: 25888648 Free PMC article. Review.
-
ApoE-associated modulation of neuroprotection from Aβ-mediated neurodegeneration in transgenic Caenorhabditis elegans.Dis Model Mech. 2019 Feb 15;12(2):dmm037218. doi: 10.1242/dmm.037218. Dis Model Mech. 2019. PMID: 30683808 Free PMC article.
-
Genome-wide screen identifies curli amyloid fibril as a bacterial component promoting host neurodegeneration.Proc Natl Acad Sci U S A. 2021 Aug 24;118(34):e2106504118. doi: 10.1073/pnas.2106504118. Proc Natl Acad Sci U S A. 2021. PMID: 34413194 Free PMC article.
-
Cellular prion protein (PrP(C)) and its role in stress responses.Int J Clin Exp Med. 2015 May 15;8(5):8042-50. eCollection 2015. Int J Clin Exp Med. 2015. PMID: 26221369 Free PMC article.
-
Protein mimetic 2D FAST rescues alpha synuclein aggregation mediated early and post disease Parkinson's phenotypes.Nat Commun. 2024 Apr 30;15(1):3658. doi: 10.1038/s41467-024-47980-4. Nat Commun. 2024. PMID: 38688913 Free PMC article.
References
-
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. - PubMed
-
- Tanner CM. Is the cause of Parkinson's disease environmental or hereditary? Evidence from twin studies. Adv Neurol. 2003;91:133–142. - PubMed
-
- Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson's disease: a meta-analysis. Environ Res. 2001;86:122–127. - PubMed
-
- Liou HH, Tsai MC, Chen CJ. Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology. 1997;48:1583–1588. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
