

Ancestral resurrection of the Drosophila S2E enhancer reveals accessible evolutionary paths through compensatory change
- PMID: 24408913
- PMCID: PMC3969564
- DOI: 10.1093/molbev/msu042
Ancestral resurrection of the Drosophila S2E enhancer reveals accessible evolutionary paths through compensatory change
Abstract
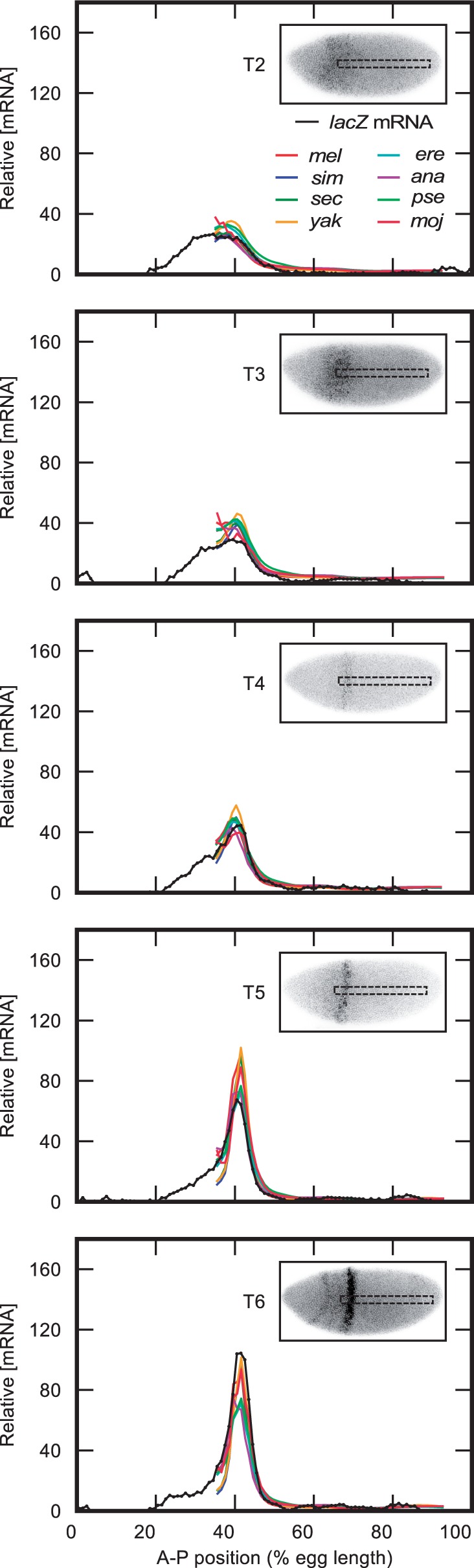
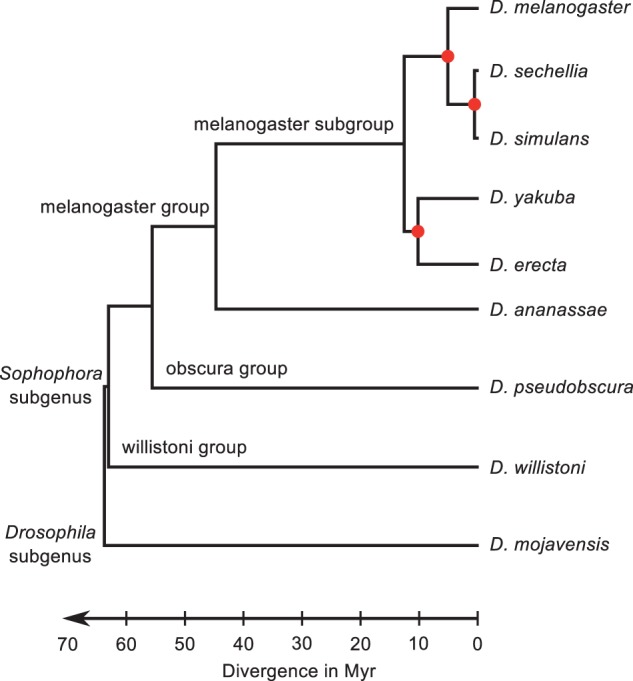
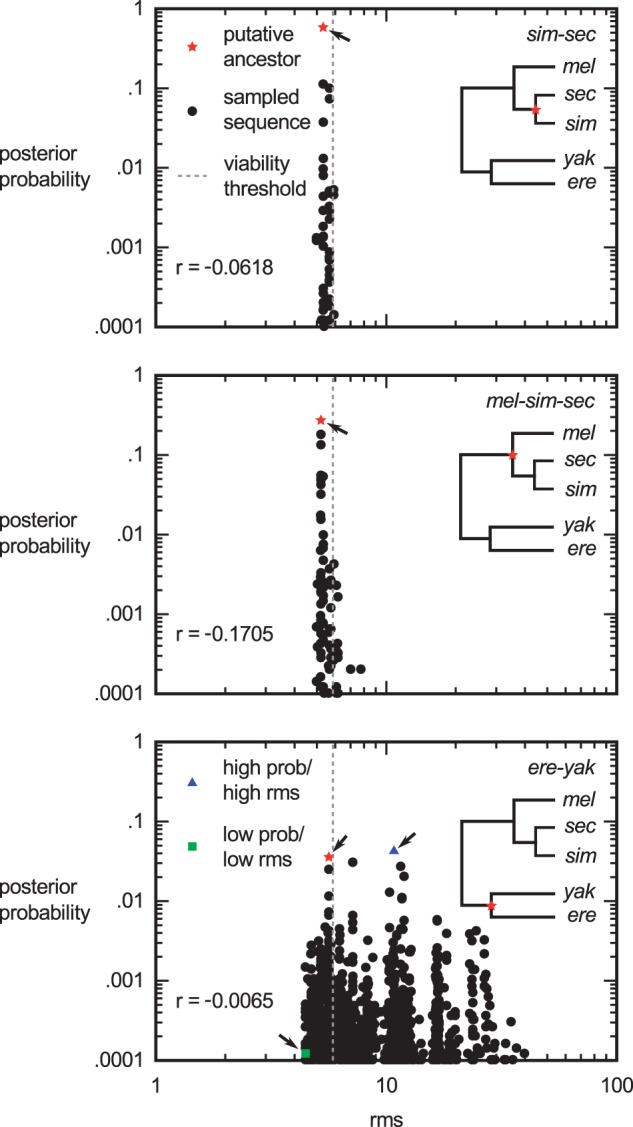
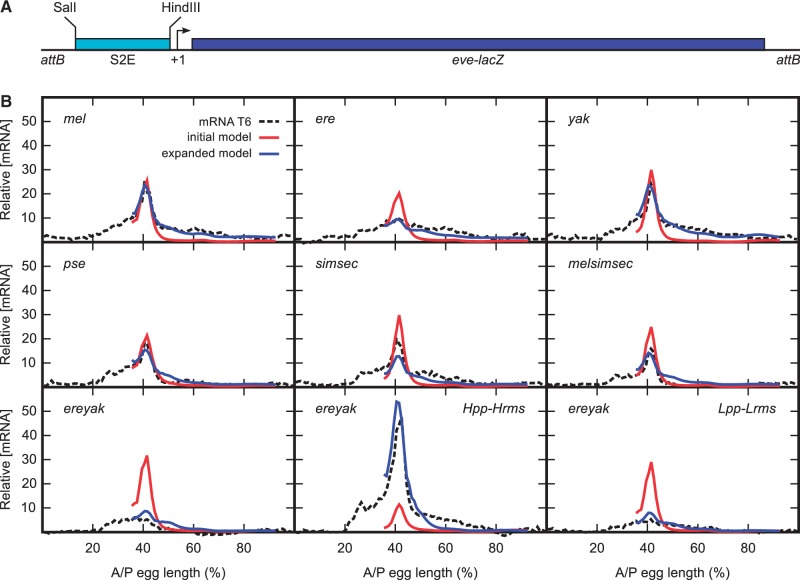
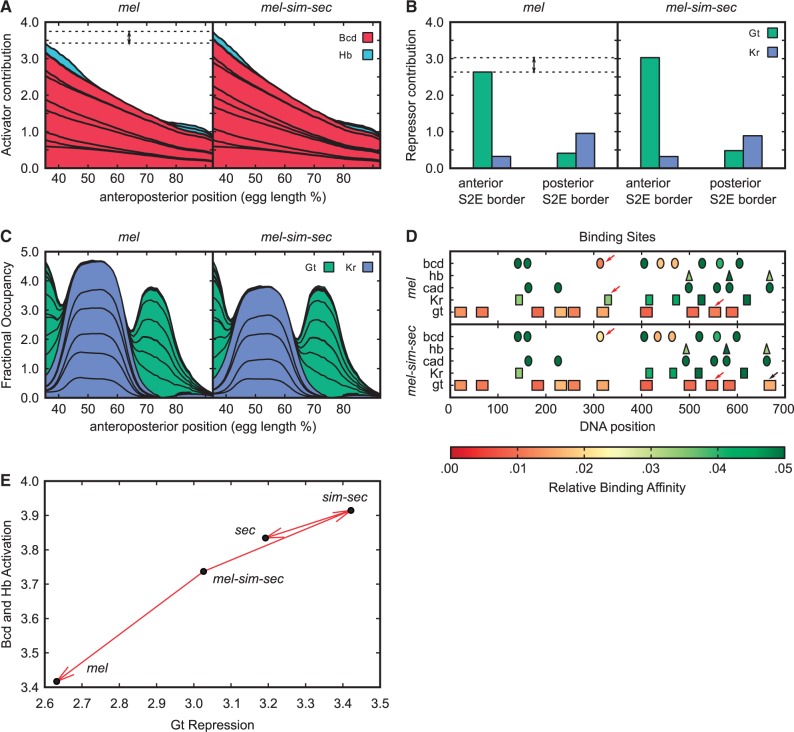
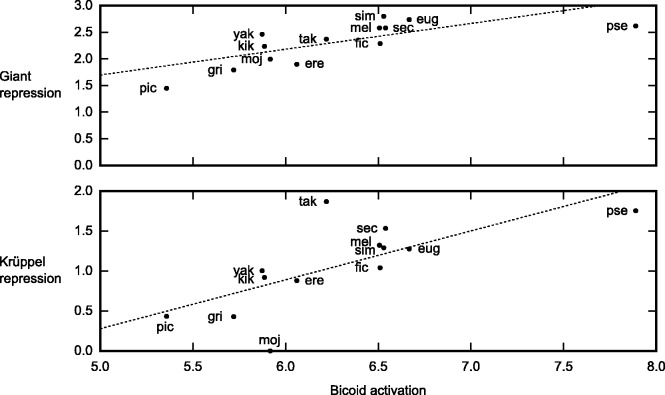
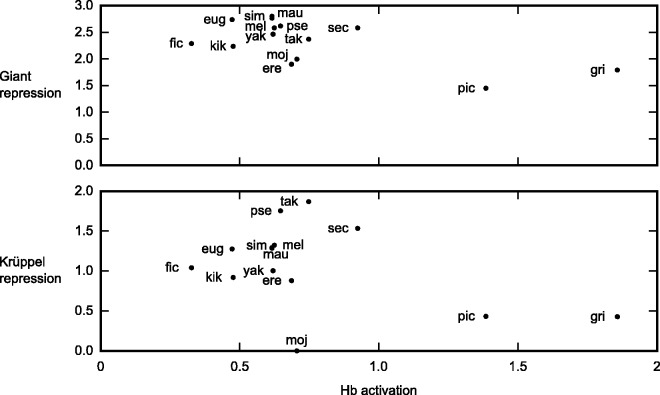
Upstream regulatory sequences that control gene expression evolve rapidly, yet the expression patterns and functions of most genes are typically conserved. To address this paradox, we have reconstructed computationally and resurrected in vivo the cis-regulatory regions of the ancestral Drosophila eve stripe 2 element and evaluated its evolution using a mathematical model of promoter function. Our feed-forward transcriptional model predicts gene expression patterns directly from enhancer sequence. We used this functional model along with phylogenetics to generate a set of possible ancestral eve stripe 2 sequences for the common ancestors of 1) D. simulans and D. sechellia; 2) D. melanogaster, D. simulans, and D. sechellia; and 3) D. erecta and D. yakuba. These ancestral sequences were synthesized and resurrected in vivo. Using a combination of quantitative and computational analysis, we find clear support for functional compensation between the binding sites for Bicoid, Giant, and Krüppel over the course of 40-60 My of Drosophila evolution. We show that this compensation is driven by a coupling interaction between Bicoid activation and repression at the anterior and posterior border necessary for proper placement of the anterior stripe 2 border. A multiplicity of mechanisms for binding site turnover exemplified by Bicoid, Giant, and Krüppel sites, explains how rapid sequence change may occur while maintaining the function of the cis-regulatory element.
Keywords: enhancer; evolution; modeling; transcription.
Figures
References
-
- Berg G, von Hippel PH. Selection of DNA binding sites by regulatory proteins. Statistical-mechanical theory and application to operators and promoters. J Mol Biol. 1987;193:723–750. - PubMed
-
- Berman BP, Pfeiffer BD, Laverty TR, Salzberg SL, Rubin GM, Eisen MB, Celniker SE. Computational identification of developmental enhancers: conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 2004;5:R61. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
