

Conformational Dynamics of the Partially Disordered Yeast Transcription Factor GCN4
- PMID: 24409105
- PMCID: PMC3882080
- DOI: 10.1021/ct400654r
Conformational Dynamics of the Partially Disordered Yeast Transcription Factor GCN4
Abstract
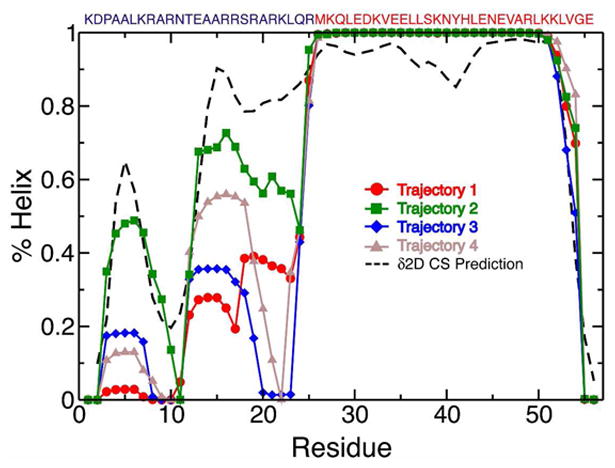
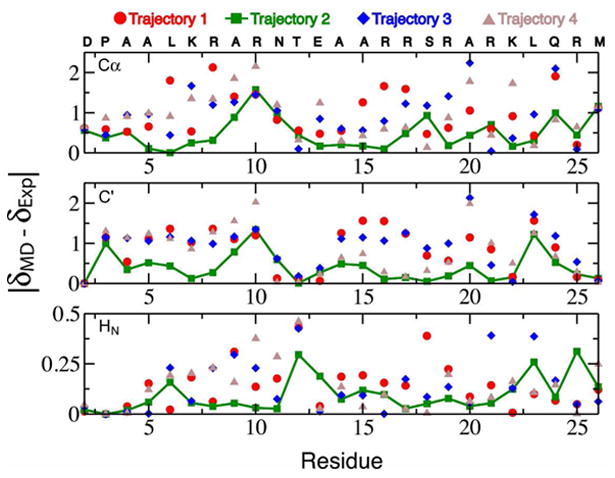
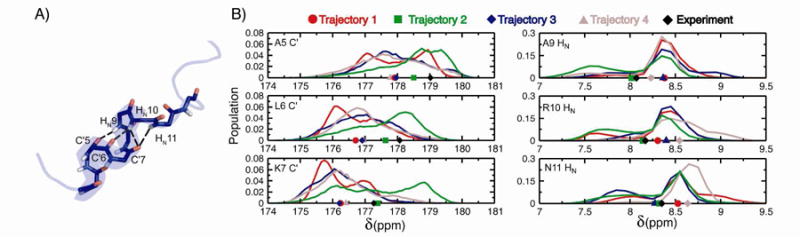
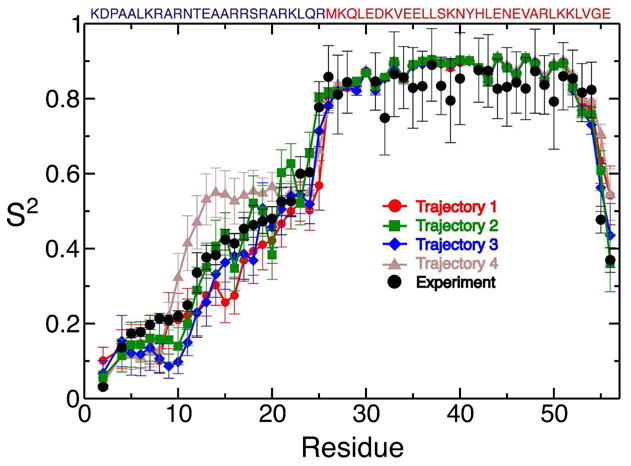
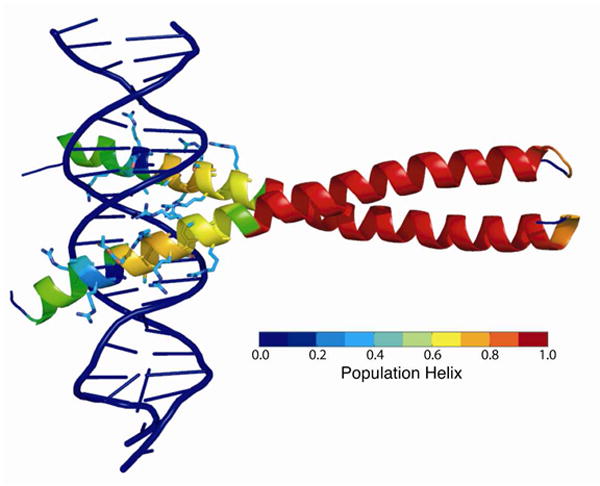
Molecular dynamics (MD) simulations have been employed to study the conformational dynamics of the partially disordered DNA binding basic leucine zipper domain of the yeast transcription factor GCN4. We demonstrate that back-calculated NMR chemical shifts and spin-relaxation data provide complementary probes of the structure and dynamics of disordered protein states and enable comparisons of the accuracy of multiple MD trajectories. In particular, back-calculated chemical shifts provide a sensitive probe of the populations of residual secondary structure elements and helix capping interactions, while spin-relaxation calculations are sensitive to a combination of dynamic and structural factors. Back calculated chemical shift and spin-relaxation data can be used to evaluate the populations of specific interactions in disordered states and identify regions of the phase space that are inconsistent with experimental measurements. The structural interactions that favor and disfavor helical conformations in the disordered basic region of the GCN4 bZip domain were analyzed in order to assess the implications of the structure and dynamics of the apo form for the DNA binding mechanism. The structural couplings observed in these experimentally validated simulations are consistent with a mechanism where the binding of a preformed helical interface would induce folding in the remainder of the protein, supporting a hybrid conformational selection / induced folding binding mechanism.
Figures
Similar articles
-
Dynamics of GCN4 facilitate DNA interaction: a model-free analysis of an intrinsically disordered region.Phys Chem Chem Phys. 2016 Feb 17;18(8):5839-49. doi: 10.1039/c5cp06197k. Phys Chem Chem Phys. 2016. PMID: 26661739 Free PMC article.
-
Temperature dependence of intramolecular dynamics of the basic leucine zipper of GCN4: implications for the entropy of association with DNA.J Mol Biol. 1999 Feb 5;285(5):2133-46. doi: 10.1006/jmbi.1998.2429. J Mol Biol. 1999. PMID: 9925790
-
A Switch between Two Intrinsically Disordered Conformational Ensembles Modulates the Active Site of a Basic-Helix-Loop-Helix Transcription Factor.J Phys Chem Lett. 2020 Nov 5;11(21):8944-8951. doi: 10.1021/acs.jpclett.0c02242. Epub 2020 Oct 8. J Phys Chem Lett. 2020. PMID: 33030907 Free PMC article.
-
Enzyme dynamics from NMR spectroscopy.Acc Chem Res. 2015 Feb 17;48(2):457-65. doi: 10.1021/ar500340a. Epub 2015 Jan 9. Acc Chem Res. 2015. PMID: 25574774 Free PMC article. Review.
-
Molecular Dynamics Simulations Combined with Nuclear Magnetic Resonance and/or Small-Angle X-ray Scattering Data for Characterizing Intrinsically Disordered Protein Conformational Ensembles.J Chem Inf Model. 2019 May 28;59(5):1743-1758. doi: 10.1021/acs.jcim.8b00928. Epub 2019 Mar 18. J Chem Inf Model. 2019. PMID: 30840442 Review.
Cited by
-
Conformational Dynamics and Allostery in E2:E3 Interactions Drive Ubiquitination: gp78 and Ube2g2.Structure. 2017 May 2;25(5):794-805.e5. doi: 10.1016/j.str.2017.03.016. Epub 2017 Apr 20. Structure. 2017. PMID: 28434917 Free PMC article.
-
What Drives 15N Spin Relaxation in Disordered Proteins? Combined NMR/MD Study of the H4 Histone Tail.Biophys J. 2018 Dec 18;115(12):2348-2367. doi: 10.1016/j.bpj.2018.11.017. Epub 2018 Nov 20. Biophys J. 2018. PMID: 30527335 Free PMC article.
-
Molecular Basis of Small-Molecule Binding to α-Synuclein.J Am Chem Soc. 2022 Feb 16;144(6):2501-2510. doi: 10.1021/jacs.1c07591. Epub 2022 Feb 7. J Am Chem Soc. 2022. PMID: 35130691 Free PMC article.
-
The transition state for coupled folding and binding of a disordered DNA binding domain resembles the unbound state.Nucleic Acids Res. 2024 Oct 28;52(19):11822-11837. doi: 10.1093/nar/gkae794. Nucleic Acids Res. 2024. PMID: 39315703 Free PMC article.
-
Fine-Tuning of ATF4 DNA Binding Activity by a Secondary Basic Motif Unique to the ATF-X Subfamily of bZip Transcription Factors.Biochemistry. 2025 Mar 18;64(6):1257-1265. doi: 10.1021/acs.biochem.4c00640. Epub 2025 Feb 24. Biochemistry. 2025. PMID: 39993237 Free PMC article.
References
-
- Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z. Biochemistry. 2002;41:6573. - PubMed
-
- Tompa P. Curr Opin Struct Biol. 2011;21:419. - PubMed
-
- Dyson HJ, Wright PE. Nature Rev Mol Cell Biol. 2005;6:197. - PubMed
-
- Dunker AK, Silman I, Uversky VN, Sussman JL. Curr Opin Struct Biol. 2008;18:756. - PubMed
-
- Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases