

Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov
- PMID: 24409173
- PMCID: PMC3873509
- DOI: 10.3389/fmicb.2013.00414
Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov
Erratum in
-
Corrigendum: Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov.Front Microbiol. 2014 Nov 4;5:583. doi: 10.3389/fmicb.2014.00583. eCollection 2014. Front Microbiol. 2014. PMID: 25406895 Free PMC article.
Abstract
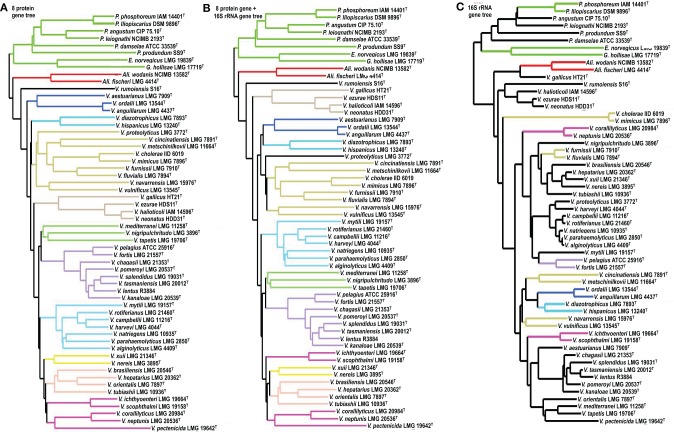
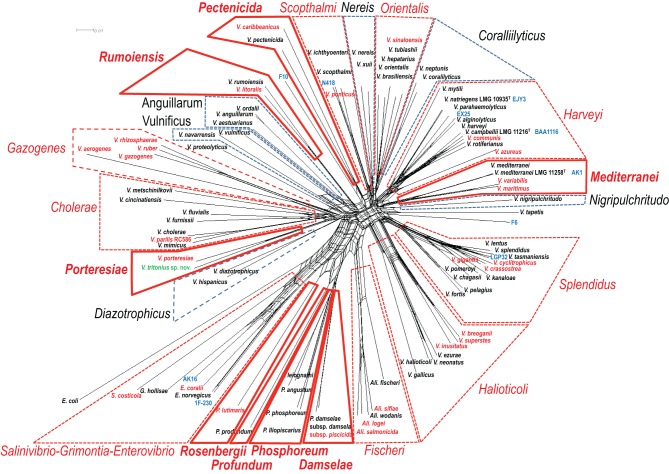
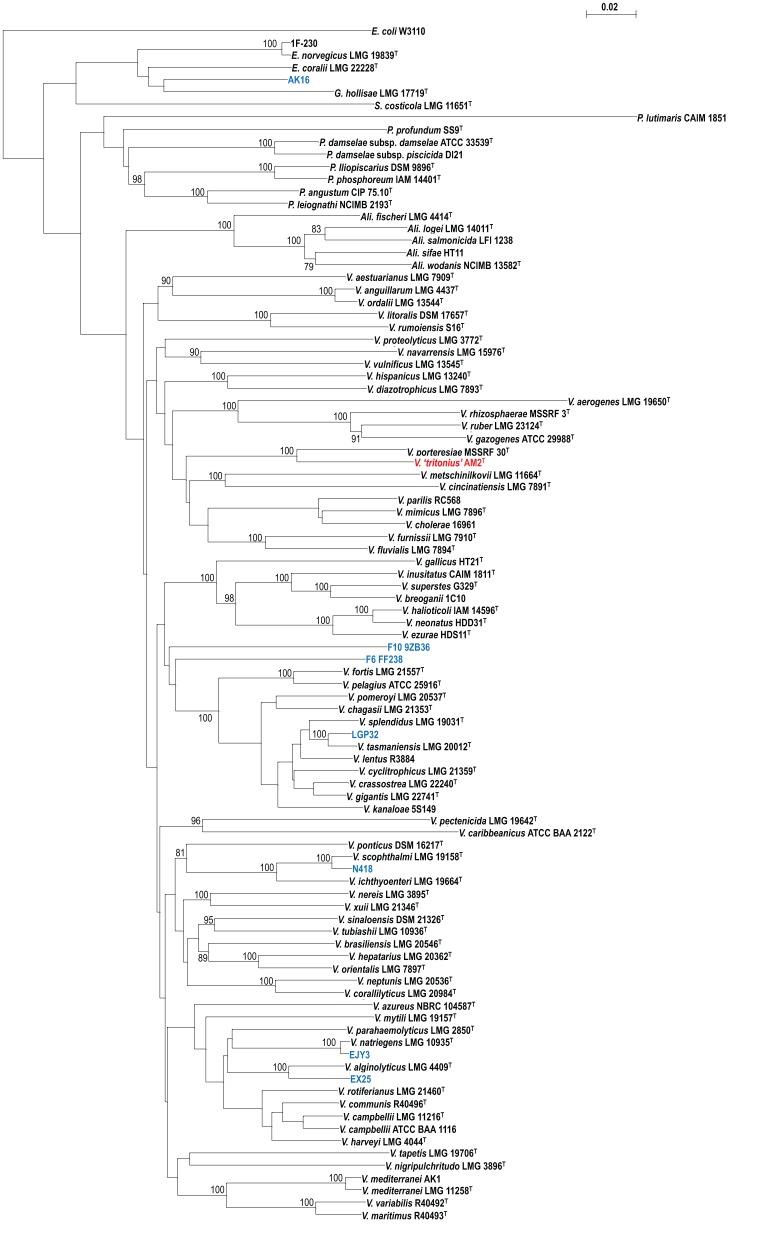
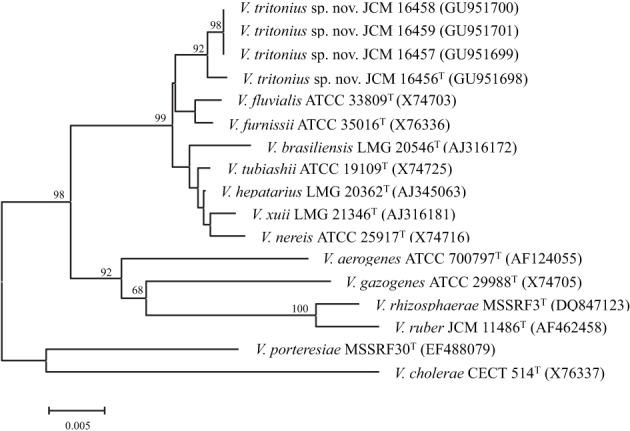
To date 142 species have been described in the Vibrionaceae family of bacteria, classified into seven genera; Aliivibrio, Echinimonas, Enterovibrio, Grimontia, Photobacterium, Salinivibrio and Vibrio. As vibrios are widespread in marine environments and show versatile metabolisms and ecologies, these bacteria are recognized as one of the most diverse and important marine heterotrophic bacterial groups for elucidating the correlation between genome evolution and ecological adaptation. However, on the basis of 16S rRNA gene phylogeny, we could not find any robust monophyletic lineages in any of the known genera. We needed further attempts to reconstruct their evolutionary history based on multilocus sequence analysis (MLSA) and/or genome wide taxonomy of all the recognized species groups. In our previous report in 2007, we conducted the first broad multilocus sequence analysis (MLSA) to infer the evolutionary history of vibrios using nine housekeeping genes (the 16S rRNA gene, gapA, gyrB, ftsZ, mreB, pyrH, recA, rpoA, and topA), and we proposed 14 distinct clades in 58 species of Vibrionaceae. Due to the difficulty of designing universal primers that can amplify the genes for MLSA in every Vibrionaceae species, some clades had yet to be defined. In this study, we present a better picture of an updated molecular phylogeny for 86 described vibrio species and 10 genome sequenced Vibrionaceae strains, using 8 housekeeping gene sequences. This new study places special emphasis on (1) eight newly identified clades (Damselae, Mediterranei, Pectenicida, Phosphoreum, Profundum, Porteresiae, Rosenbergii, and Rumoiensis); (2) clades amended since the 2007 proposal with recently described new species; (3) orphan clades of genomospecies F6 and F10; (4) phylogenetic positions defined in 3 genome-sequenced strains (N418, EX25, and EJY3); and (5) description of V. tritonius sp. nov., which is a member of the "Porteresiae" clade.
Keywords: Vibrio tritonius; Vibrionaceae; evolution; housekeeping protein gene; multilocus sequence analysis; vibrios.
Figures
References
-
- Aujoulat F., Jumas-Bilak E., Masnou A., Sallé F., Faure D., Segonds C., et al. (2011). Multilocus sequence-based analysis delineates a clonal population of Agrobacterium (Rhizobium) radiobacter (Agrobacterium tumefaciens) of human origin. J. Bacteriol. 193, 2608–2618 10.1128/JB.00107-11 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous