

Targeting heat shock proteins to modulate α-synuclein toxicity
- PMID: 24409201
- PMCID: PMC3886379
- DOI: 10.1177/1756285613493469
Targeting heat shock proteins to modulate α-synuclein toxicity
Abstract
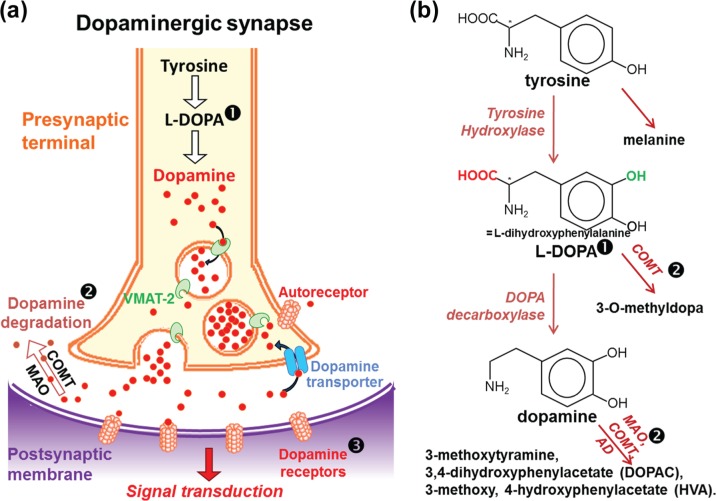
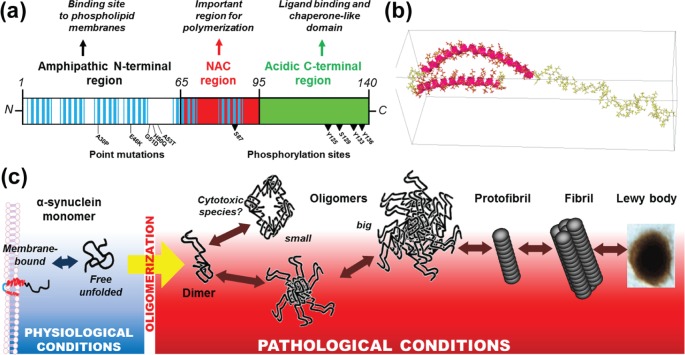
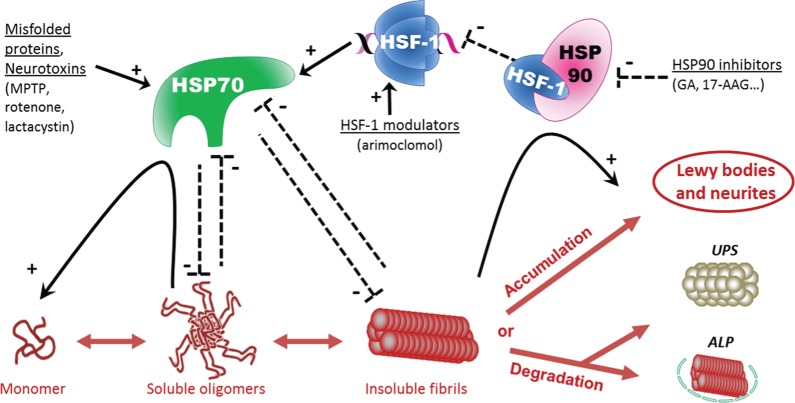
Parkinson's disease is a slowly progressive neurodegenerative disorder typically characterized by the loss of dopaminergic neurons within the substantia nigra pars compacta, and the intraneuronal deposition of insoluble protein aggregates chiefly comprised of α-synuclein. Patients experience debilitating symptoms including bradykinesia, rigidity and postural instability. No curative treatment currently exists and therapeutic strategies are restricted to symptomatic treatment only. Over the past decade a class of molecular chaperones called the heat shock proteins has emerged as a potentially promising therapeutic target. Heat shock proteins aid in the folding and refolding of proteins, and target denatured proteins to degradation systems. By targeting heat shock proteins through various means including overexpression and pharmacological enhancement, researchers have shown that α-synuclein aggregation and its associated cytotoxicity can be therapeutically modulated in an array of cell and animal models. This review highlights the relevant progress in this field and discusses the relevance of heat shock proteins as therapeutic modulators of α-synuclein toxicity to the rapidly evolving understanding of Parkinson's disease pathogenesis.
Keywords: heat shock protein; molecular chaperones; parkinsonism; α-synuclein.
Conflict of interest statement
Figures
References
-
- Abeliovich A., Schmitz Y., Farinas I., Choi-Lundberg D., Ho W., Castillo P., et al. (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: 239–252 - PubMed
-
- Alcain F., Villalba J. (2009) Sirtuin activators. Expert Opin Ther Pat 19: 403–414 - PubMed
-
- Al-Ramahi I., Lam Y., Chen H., De Gouyon B., Zhang M., Perez A., et al. (2006) CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 281: 26714–26724 - PubMed
-
- Anfinsen C. (1973) Principles that govern the folding of protein chains. Science 181: 223–230 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
