

High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion
- PMID: 24410116
- PMCID: PMC3985945
- DOI: 10.1021/ja411119m
High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion
Abstract
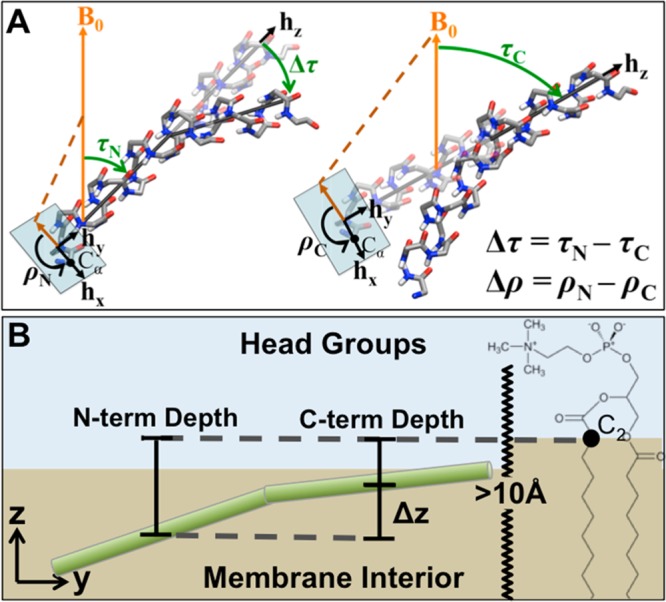
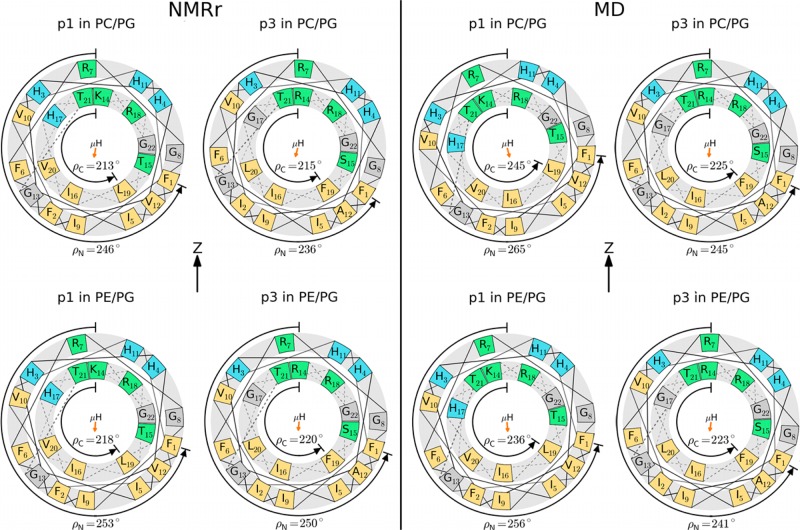
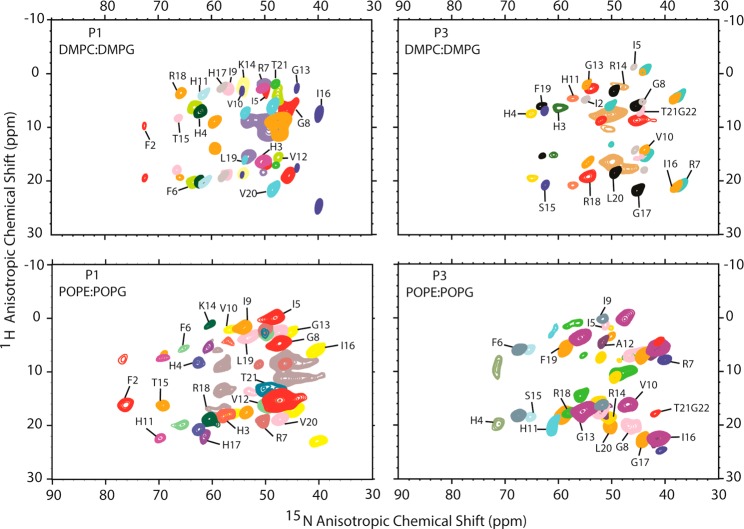
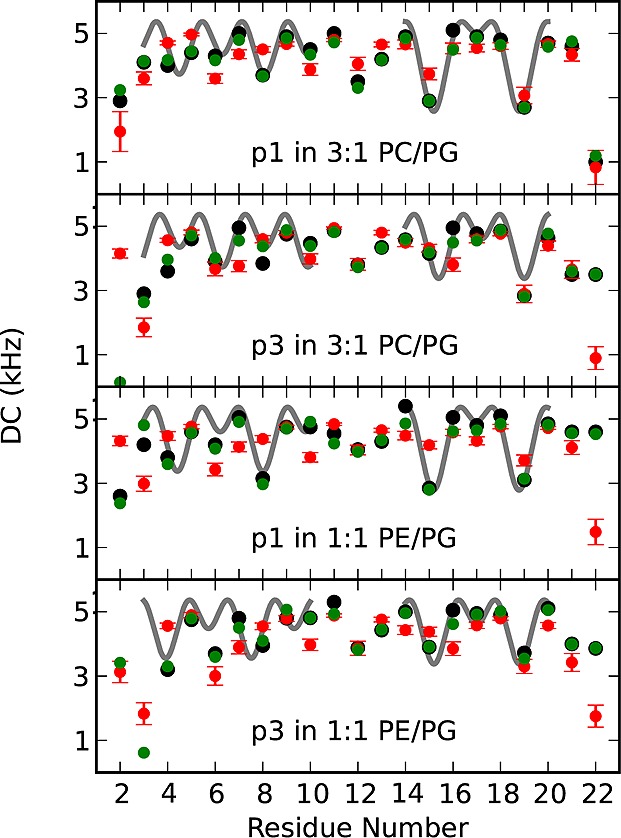
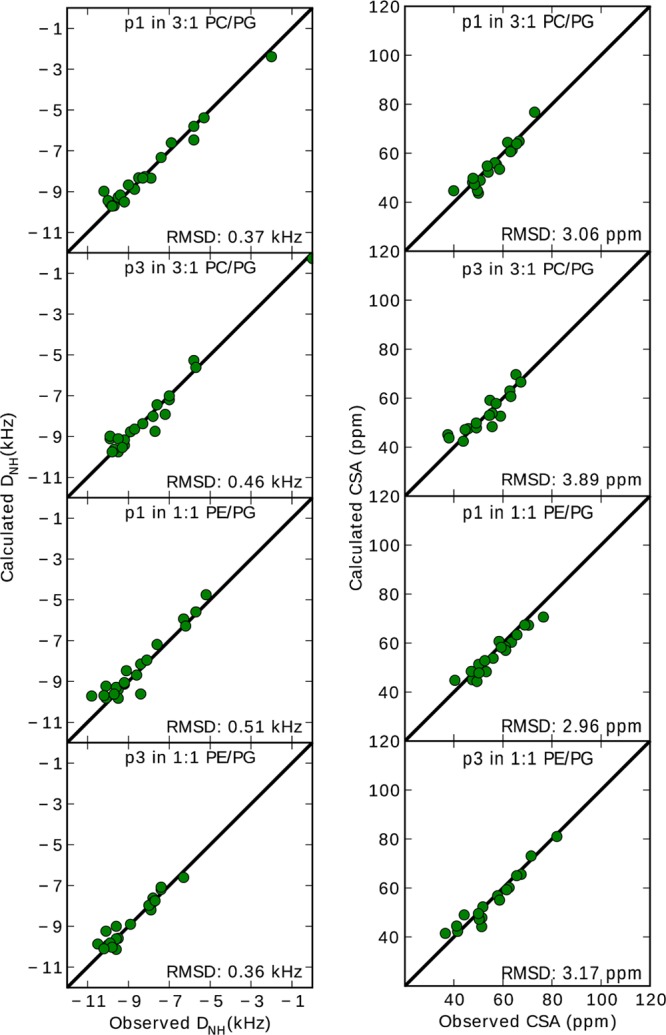
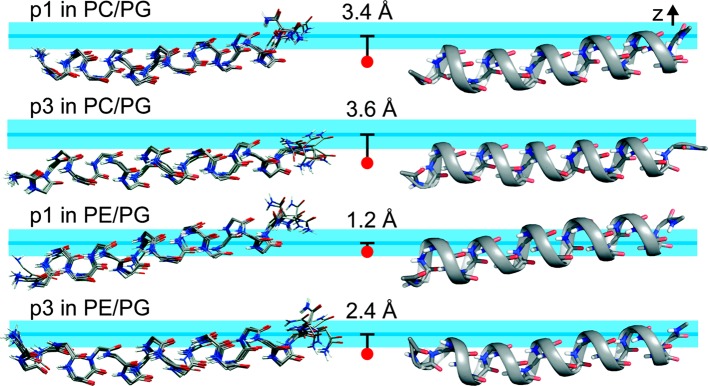
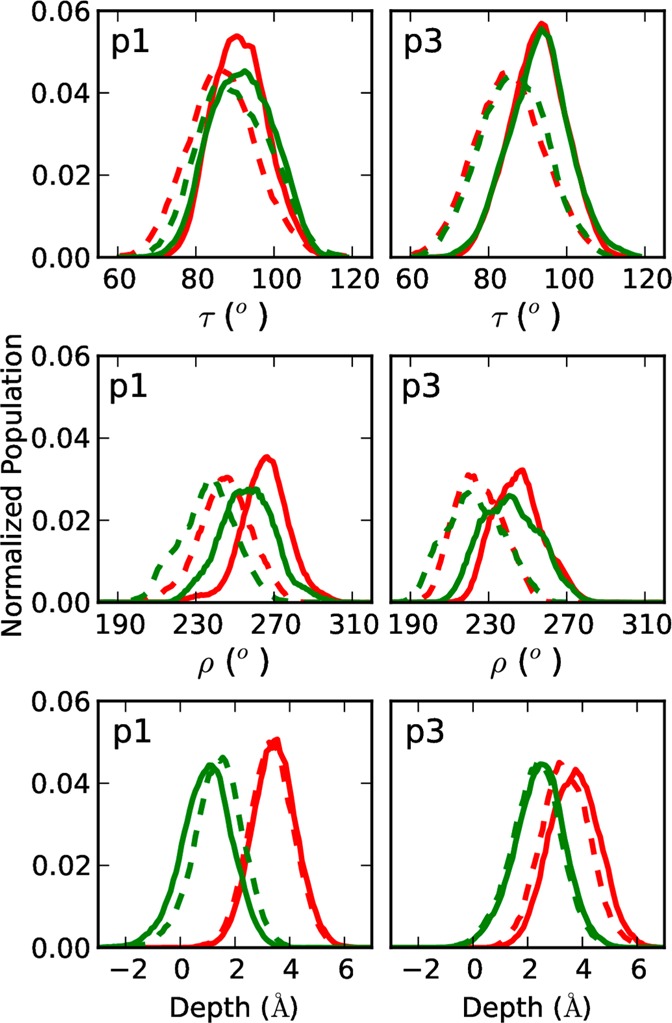
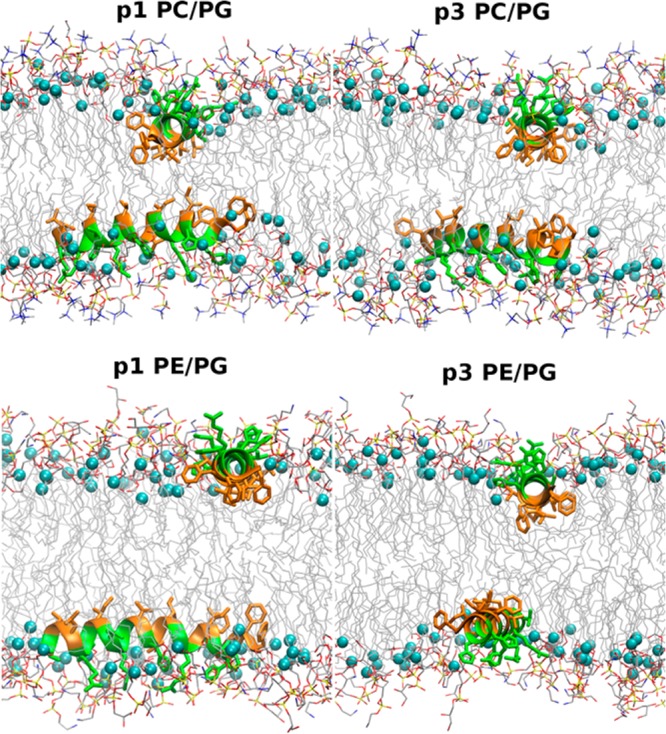
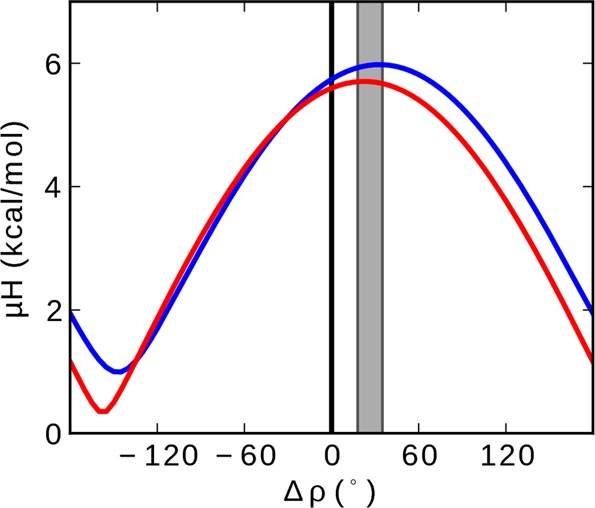
While antimicrobial peptides (AMPs) have been widely investigated as potential therapeutics, high-resolution structures obtained under biologically relevant conditions are lacking. Here, the high-resolution structures of the homologous 22-residue long AMPs piscidin 1 (p1) and piscidin 3 (p3) are determined in fluid-phase 3:1 phosphatidylcholine/phosphatidylglycerol (PC/PG) and 1:1 phosphatidylethanolamine/phosphatidylglycerol (PE/PG) bilayers to identify molecular features important for membrane destabilization in bacterial cell membrane mimics. Structural refinement of (1)H-(15)N dipolar couplings and (15)N chemical shifts measured by oriented sample solid-state NMR and all-atom molecular dynamics (MD) simulations provide structural and orientational information of high precision and accuracy about these interfacially bound α-helical peptides. The tilt of the helical axis, τ, is between 83° and 93° with respect to the bilayer normal for all systems and analysis methods. The average azimuthal rotation, ρ, is 235°, which results in burial of hydrophobic residues in the bilayer. The refined NMR and MD structures reveal a slight kink at G13 that delineates two helical segments characterized by a small difference in their τ angles (<10°) and significant difference in their ρ angles (~25°). Remarkably, the kink, at the end of a G(X)4G motif highly conserved among members of the piscidin family, allows p1 and p3 to adopt ρ angles that maximize their hydrophobic moments. Two structural features differentiate the more potent p1 from p3: p1 has a larger ρ angle and less N-terminal fraying. The peptides have comparable depths of insertion in PC/PG, but p3 is 1.2 Å more deeply inserted than p1 in PE/PG. In contrast to the ideal α-helical structures typically assumed in mechanistic models of AMPs, p1 and p3 adopt disrupted α-helical backbones that correct for differences in the amphipathicity of their N- and C-ends, and their centers of mass lie ~1.2-3.6 Å below the plane defined by the C2 atoms of the lipid acyl chains.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
