

Deletion of a conserved cis-element in the Ifng locus highlights the role of acute histone acetylation in modulating inducible gene transcription
- PMID: 24415943
- PMCID: PMC3886902
- DOI: 10.1371/journal.pgen.1003969
Deletion of a conserved cis-element in the Ifng locus highlights the role of acute histone acetylation in modulating inducible gene transcription
Abstract
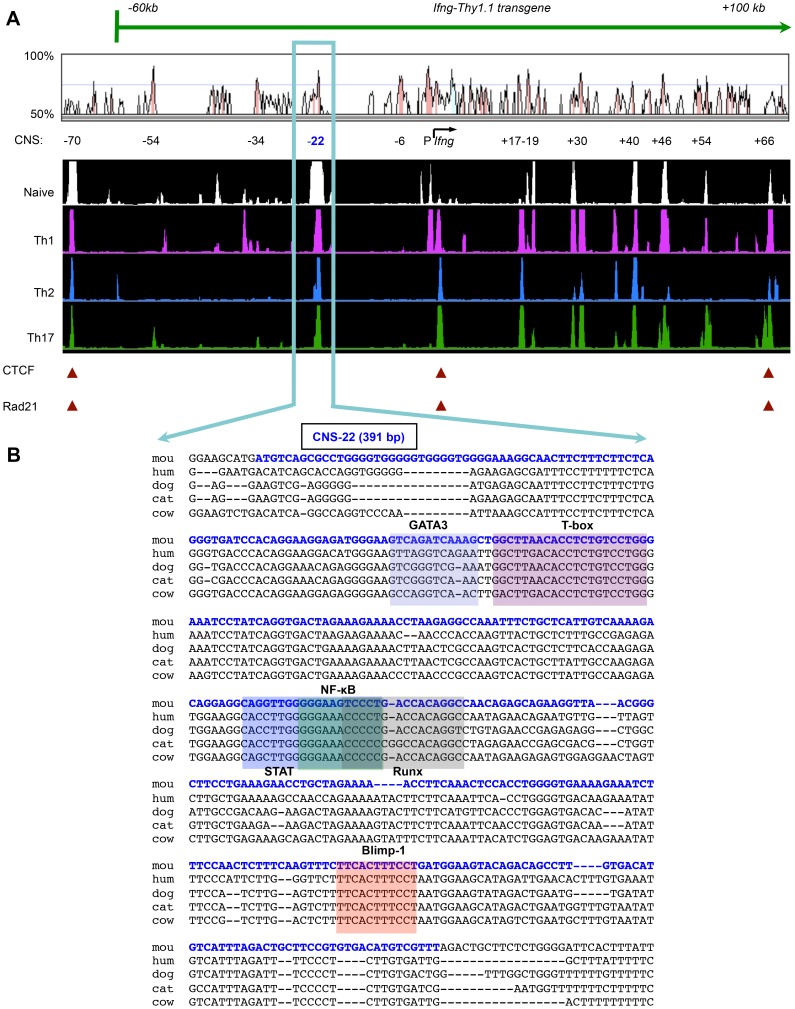
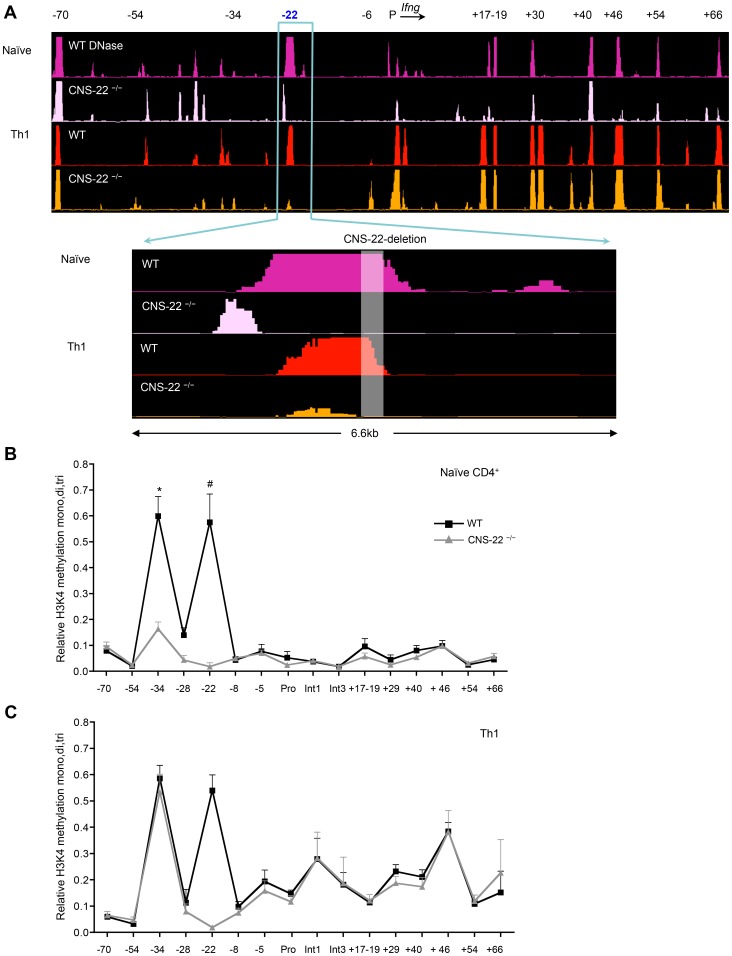
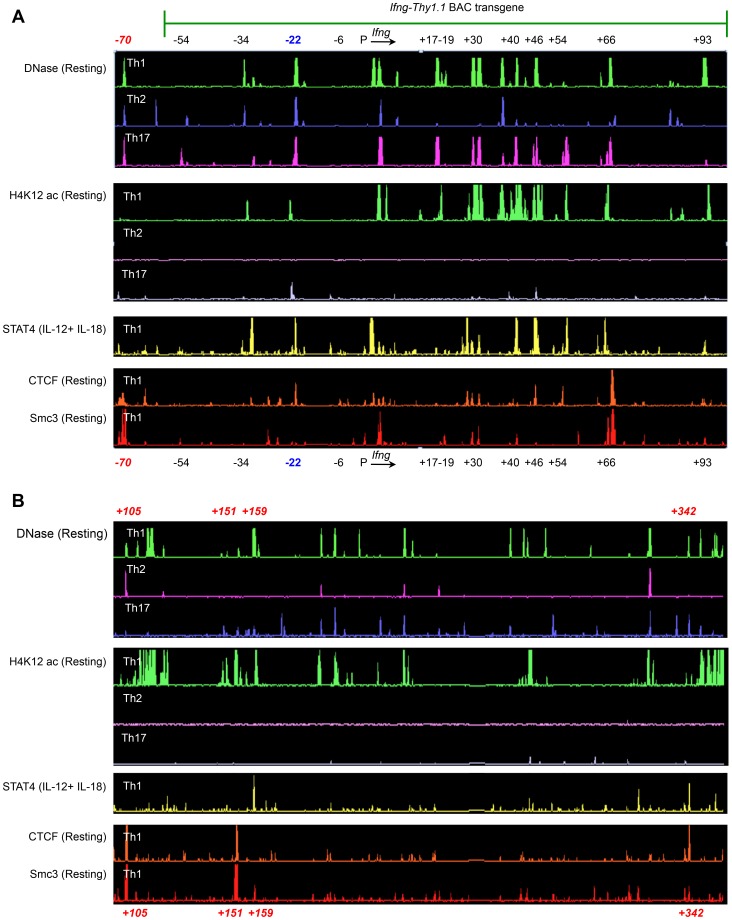
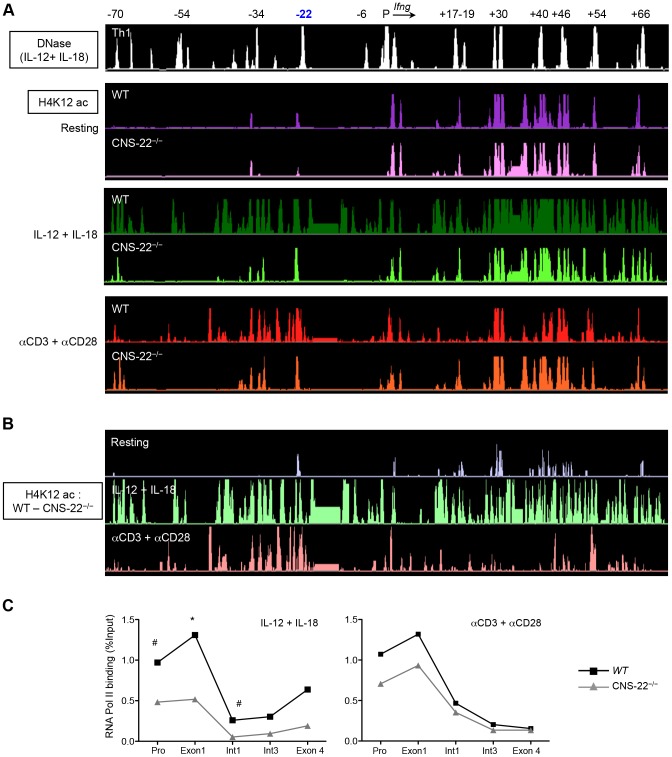
Differentiation-dependent regulation of the Ifng cytokine gene locus in T helper (Th) cells has emerged as an excellent model for functional study of distal elements that control lineage-specific gene expression. We previously identified a cis-regulatory element located 22 kb upstream of the Ifng gene (Conserved Non-coding Sequence -22, or CNS-22) that is a site for recruitment of the transcription factors T-bet, Runx3, NF-κB and STAT4, which act to regulate transcription of the Ifng gene in Th1 cells. Here, we report the generation of mice with a conditional deletion of CNS-22 that has enabled us to define the epigenetic and functional consequences of its absence. Deletion of CNS-22 led to a defect in induction of Ifng by the cytokines IL-12 and IL-18, with a more modest effect on induction via T-cell receptor activation. To better understand how CNS-22 and other Ifng CNSs regulated Ifng transcription in response to these distinct stimuli, we examined activation-dependent changes in epigenetic modifications across the extended Ifng locus in CNS-22-deficient T cells. We demonstrate that in response to both cytokine and TCR driven activation signals, CNS-22 and other Ifng CNSs recruit increased activity of histone acetyl transferases (HATs) that transiently enhance levels of histones H3 and H4 acetylation across the extended Ifng locus. We also demonstrate that activation-responsive increases in histone acetylation levels are directly linked to the ability of Ifng CNSs to acutely enhance Pol II recruitment to the Ifng promoter. Finally, we show that impairment in IL-12+IL-18 dependent induction of Ifng stems from the importance of CNS-22 in coordinating locus-wide levels of histone acetylation in response to these cytokines. These findings identify a role for acute histone acetylation in the enhancer function of distal conserved cis-elements that regulate of Ifng gene expression.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
